


0 引言
【研究意义】理想株型可以提高玉米种植密度和光合效率,是玉米高产的主要因素[1]。叶宽是玉米株型的主要性状之一,对其进行遗传分析,对选育高产耐密玉米品种具有重要意义[2]。【前人研究进展】Ku等[3]用玉米自交系Yu82和Shen137组配了含有229个家系的F2∶3群体,在第1、4、7和8号染色体上,共检测到5个与叶宽相关的QTL,可以解释34.13%的表型变异。唐登国[4]用208个分子标记在B73和1212组配的325个重组自交系群体对叶片性状进行QTL定位,结果显示控制叶宽的QTL大多在4、5、6和8号染色体上。张莹莹等[5]以郑58和D863F构建RIL群体,在5号和8号染色体上,定位到5个穗位叶宽QTL,单个QTL表型贡献率8.35%~12.34%。张旷野[6]利用自交系掖478和齐319组配RIL群体,结合152个SSR标记,对多环境下玉米穗位叶宽进行QTL定位,发现20个QTL位点,其中有7个主效QTL。王会涛等[7]以豫82和豫87-1为材料构建RIL群体,共检测到7个与玉米穗位叶宽相关的QTL,分布在第1、2、3、6、8和10号染色体上。全基因组选择(genomic selection, GS)是利用训练群体的基因型和表型数据建模的一种育种手段,对只有基因型的育种群体进行表型预测和选择。常规育种基于性状表型值,通过BLUP估算育种值(EBVs),而GS则通过全基因组中单个标记效应值的累加,获得基因组估计育种值(GEBV)[8]。Bernardo[9]使用外国玉米与本地玉米杂交,组配多种群体进行全基因组选择,结果表明全基因组选择应该从F2群体开始较为合适。Zhao等[10]使用6个家系的双亲后代群体进行测交获得表型值,结合SNP标记进行全基因组选择,发现SNP标记数量从100开始逐渐上升后,预测精度也逐渐上升,标记数量达到800个SNP后预测精度达到稳定水平。【本研究切入点】虽然目前已经定位到了控制玉米叶宽的QTL。但由于作图群体、种植环境、遗传标记不同,检测到的QTL一致性较差,所以挖掘在多环境下稳定存在的QTL对分子标记辅助选择育种有重要意义[11-12]。【拟解决的关键问题】以郑58和B73为亲本构建F2∶3作图群体,在多环境下表型测量玉米穗位叶宽,采用液相48K探针捕获技术进行基因型检测,对玉米叶宽表型进行QTL定位,获得控制叶宽的主效QTL位点,并对玉米叶宽性状进行全基因组选择,研究标记数目、训练群体的大小对全基因组选择预测精度的影响,分析双亲群体全基因组选择的规律,为玉米叶宽的全基因选择提供理论依据。
1 材料与方法
1.1 材料
玉米自交系B73和郑58于2018年冬季在海南省乐东黎族自治县实验站种植并杂交获得F1,2019年夏季将F1种植于新疆昌吉市九圣禾实验站,并自交获得F2,2019年冬季将F2在海南省乐东黎族自治县实验站种植自交获得200个F2∶3家系。将F2∶3家系于2020年夏季种植于新疆昌吉市九圣禾实验站和新疆乌鲁木齐市三坪试验站,2020年冬季种植在海南省乐东黎族自治县实验站。每个地点设置2个重复,采用完全随机区组试验设计,行长3 m,株距0.25 m,行距0.67 m,单行区种植,田间管理同大田生产。在F2∶3群体玉米灌浆时期从每个小区的第5株开始连续选取5株长势均衡的单株调查,用直尺测量穗位叶的最宽处。
1.2 方法
1.2.1 表型数据
利用META-R软件(http://hdl.handle.net/11529/10201)对3个环境下的穗位叶宽表型数据进行分析,获得最佳线性无偏估计值(Best Linear Unbiased Prediction BLUP),用于QTL定位和全基因组选择分析。利用QTL IciMapping软件AOV(ANOVA OF multi-environmental trials)功能进行方差分析,计算遗传力[13]:
式中,H2代表广义遗传力,VG代表基因型方差,VGE代表基因环境相互作用方差,Vε代表误差方差,e代表环境,r代表重复。
1.2.2 基因分型和连锁图谱构建
将SNP标记根据物理位置进行排序后导入QTL IciMapping软件,选择MAP功能进行连锁群的划分及遗传距离的确定,最终获得总长度为876.73 cM,相邻标记间的距离为0.49 cM的连锁图谱。
1.2.3 QTL定位
采用META-R软件获得的BLUP值作为表型值,使用QTL IciMapping 4.2软件的BIP功能对三个环境下及联合BLUP的穗位叶宽性状进行QTL定位,定位方法采用复合区间作图加性模型(ICIM-ADD)。分子标记之间每隔0.5 cM进行一次全基因组扫描,窗口的设定为5 cM,将LOD值设定为2.5。以该位点的显性效应与加性效应比值的绝对值作为显性度,对QTL位点的遗传作用方式进行分类。显性度在(0~0.20)区间则为加性(A);显性度在(0.21~0.80)区间则为部分显性(PD);显性度在(0.81~1.20)则为显性(D);当显性度大于1.20是表示超显性(OD)[16]。
1.2.4 全基因组选择
根据液相48K探针捕获技术获得的62 504个SNP标记作为基因组,多环境的BLUP值作为表型值,使用R软件RR-BLUP包对穗位叶宽性状进行全基因组选择分析。采用k=5的K-fold交叉验证进行比较,随机选取群体的20%作为预测群体,剩下的80%作为训练群体,进行全基因组选择,全基因组选择预测精度通过预测群体的真实育种值与全基因组估计育种值间的相关系数计算,重复100次。
SNP标记个数设定为10、30、50、100、300、500、1 000、3 000、5 000、10 000和50 000个,采用五折交叉验证方法,每个标记密度进行100次全基因组选择,研究标记数目对全基因组选择预测精度的影响。训练群体大小设定为总群体的10%、20%、30%、40%、50%、60%、70%、80%和90%,剩余群体为预测群体,采用全部62 504个SNP标记,每个训练群体大小进行100次全基因组选择。
2 结果与分析
2.1 玉米叶宽表型数据
研究表明,B73叶宽的均值为9.04 cm,最小值为7.82 cm,最大值为10.32 cm;郑58叶宽的均值为8.02 cm,最小值为6.97 cm,最大值为8.89 cm。郑58叶宽比B73叶宽窄,存在极显著差异,组配的F2∶3家系可以用于玉米叶宽的遗传解析。海南实验站、九圣禾实验站、三坪实验站及3个环境联合叶宽平均值分别为8.68、8.04、7.24和8.00 cm。3个地点玉米叶宽都表现出了广泛的变异,其中九圣禾实验站的变异幅度最大,变幅为1.76,标准差为0.31;海南实验站的变异幅度最小,变幅为0.27,标准差为0.06。叶宽在3个环境下的偏度和峰度的绝对值均小于1,F2∶3家系的叶宽符合正态分布。图1,表1
图1
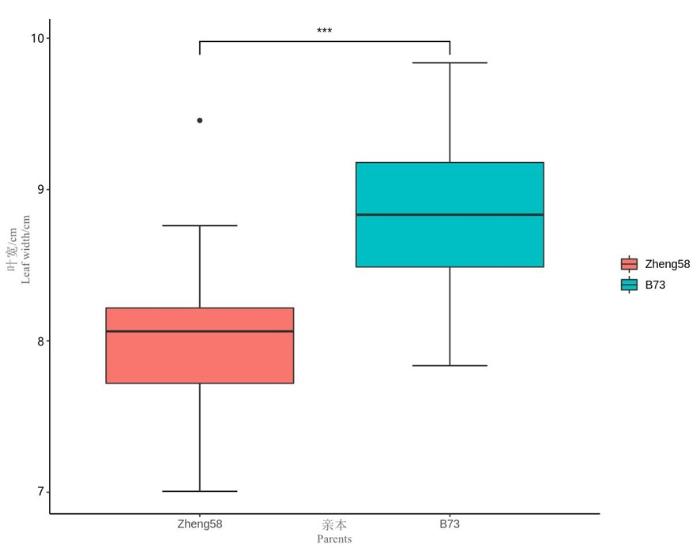
表1 亲本和F2∶3家系叶宽的描述性统计
Tab.1
| 亲本Parents | 环境 Environ- ment | 均值 Mean | 最小值 Max. | 最大值 Min. | 标准差 S.D. | 变幅 A.O.F | 变异 系数 CV. | 峰度 Kurtosis | 偏度 Skewness | |
|---|---|---|---|---|---|---|---|---|---|---|
| 郑58 | B73 | |||||||||
| 8.02 | 9.04 | 海南 | 8.68 | 8.55 | 8.82 | 0.06 | 0.27 | 0.01 | 0.21 | -0.44 |
| 九圣禾 | 8.04 | 7.23 | 8.99 | 0.31 | 1.76 | 0.04 | 0.09 | -0.22 | ||
| 三坪 | 7.24 | 6.96 | 7.63 | 0.13 | 0.67 | 0.02 | 0.07 | -0.02 | ||
| 联合 | 8.00 | 7.78 | 8.19 | 0.07 | 0.41 | 0.01 | -0.17 | -0.11 | ||
2.2 玉米叶宽方差
研究表明,环境、基因型和环境与基因型互作各个组分的P值均小于0.01,环境、基因型和环境与基因型互作对玉米叶宽表型均有极显著影响。叶宽的遗传力为0.39,遗传力较低。表2
表2 F2∶3家系叶宽方差和广义遗传力
Tab.2
| 变异来源 Source | 自由度 Degree of freedom | 平方和 Sum of square | 均方 Mean of square | F | P | 遗传力 H2 |
|---|---|---|---|---|---|---|
| 环境Environment | 2 | 420.49 | 210.24 | 726.28 | <0.01** | 0.39 |
| 基因型Genotype | 199 | 109.78 | 0.55 | 1.91 | <0.01** | |
| 基因型环境互作GXE | 398 | 166.11 | 0.42 | 1.44 | <0.01** | |
| 误差Error | 597 | 172.82 | 0.29 | |||
| 总变异Variation | 1 196 | 869.20 |
注:*表示在0.05水平上差异显著;**表示在0.01水平上差异显著
Note:*significance at P<0.05; **significance at P<0.01
2.3 玉米叶宽QTL定位
研究表明,共检测到12个控制穗位叶宽的QTL位点,在第1、3、4、5、8和10号染色体上均有分布。在海南环境中定位到4个QTL,分别位于第1、3、5和8号染色体,表型贡献率为4.41%~16.17%。其中位于5号染色体的qew5-1表型贡献率为16.17%,是主效QTL位点。三坪环境中定位到2个QTL qew1-2和qew4-1,分别在第1和4号染色体上,分别解释表型变异为5.88%和10.63%。在九圣禾环境中定位到1个QTL qew8-2,位于第8号染色体上,表型贡献率为6.28%。定位到5个QTL,分布在第1、3、4、5和10号染色体上,单个QTL表型贡献率为3.75%~13.18%。其中在海南检测到的qew1-1和联合分析检测到的qew1-3是同一个位点,位于bin 1.06,LOD值分别为2.73和4.02,表型贡献率分别为5.04%和5.47%。在海南检测到的bin 5.01的qew5-1和联合分析检测到的qew5-2是同一个位点,位于bin 5.01,LOD值分别为8.06和9.08,表型贡献率分别为16.17%和13.18%,是控制叶宽的主效QTL位点。qew1-1(qew1-3)和qew5-1(qew5-2)是多环境稳定存在的控制叶宽的QTL。
有7个位点qew3-1、qew5-1、qew1-2、qew1-3、qew3-2、qew5-2和qew10-1呈现加性效应,有两个位点qew1-1和qew4-3表现为部分显性,有一个位点qew4-1表现为完全显性,有两个位点qew8-1和qew8-2表现为超显性。玉米穗位叶宽的作用方式以加性效应为主,还有部分表现为不同程度的显性。表3
表3 F2∶3 家系玉米叶宽的QTL定位结果
Tab.3
| QTL | 环境 Environ ment | 染色体 /Bin Chromo- some /Bin | 遗传位置 Genetic position (cM) | 物理距离 /Mba Physical position /Mba | 左翼标记 Left marker | 右翼标记 Right marker | LOD值 | 表型 贡献率 PVE (%) | 加性 效应 Add | 显性 效应 Dom | 基因 效应b Gene action |
|---|---|---|---|---|---|---|---|---|---|---|---|
| qew1-1 | 海南 | 1/1.06 | 74 | 196.18~197.14 | RM1_149 | RM1_150 | 2.73 | 5.04 | 0.1 | -0.06 | PD |
| qew3-1 | 海南 | 3/3.07 | 52 | 198.91~198.92 | RM3_117 | RM3_118 | 2.93 | 5.51 | -0.1 | -0.01 | A |
| qew5-1 | 海南 | 5/5.01 | 17 | 8.86~8.90 | RM5_27 | RM5_26 | 8.06 | 16.17 | 0.2 | 0.01 | A |
| qew8-1 | 海南 | 8/8.02 | 13 | 13.54~13.64 | RM8_22 | RM8_23 | 2.52 | 4.41 | 0.06 | 0.09 | OD |
| qew1-2 | 三坪 | 1/1.07 | 84 | 212.21~212.36 | RM1_173 | RM1_174 | 2.57 | 5.88 | 0.05 | -0.01 | A |
| qew4-1 | 三坪 | 4/4.08 | 60 | 188.92~189.03 | RM4_135 | RM4_136 | 4.66 | 10.63 | 0.05 | 0.05 | D |
| qew8-2 | 九圣禾 | 8/8.05 | 31 | 138.67~138.78 | RM8_63 | RM8_64 | 2.65 | 6.28 | 0.01 | 0.02 | OD |
| qew1-3 | 联合 | 1/1.06 | 74 | 196.18~197.14 | RM1_149 | RM1_150 | 4.02 | 5.47 | 0.05 | -0.01 | A |
| qew3-2 | 联合 | 3/3.04 | 28 | 36.82~50.97 | RM3_50 | RM3_51 | 3.93 | 5.21 | -0.05 | -0.01 | A |
| qew4-2 | 联合 | 4/4.09 | 69 | 223.31~224.37 | RM4_155 | RM4_156 | 6.03 | 8.19 | 0.06 | 0.02 | PD |
| qew5-2 | 联合 | 5/5.01 | 17 | 8.86~8.90 | RM5_27 | RM5_26 | 9.08 | 13.18 | 0.08 | 0.01 | A |
| qew10-1 | 联合 | 10/10.07 | 56 | 146.71~146.77 | RM10_102 | RM10_103 | 2.78 | 3.75 | 0.04 | 0.0001 | A |
注:a基于B73_RefGen_V4_genomic参考基因组的位置;b基因作用方式:A代表加性,PD代表部分显性,D代表显性。OD代表超显性
note:aphysical position corresponds to maize B73_RefGen_v4 genomic;bGene action was determined according to:additive(A),partially dominant(PD),dominant(D), over dominant(OD)
2.4 玉米叶宽的全基因组选择
研究表明,对玉米F2群体的叶宽性状进行全基因组选择,预测精度仅为0.34。
当使用10、30、50、100、300个SNP标记对叶宽进行全基因组选择分析时,叶宽的预测精度分别在0.12、0.19、0.22、0.25、0.28附近均匀分布,叶宽预测精度分别提升了0.07、0.03、0.03、0.03。标记数目从300增加到50 000时,叶宽的预测精都在0.28处附近均匀分布,前后差异不明显。在双亲后代群体中,使用300个SNP标记进行全基因组选择可以得到比较好的预测精度。图2
图2
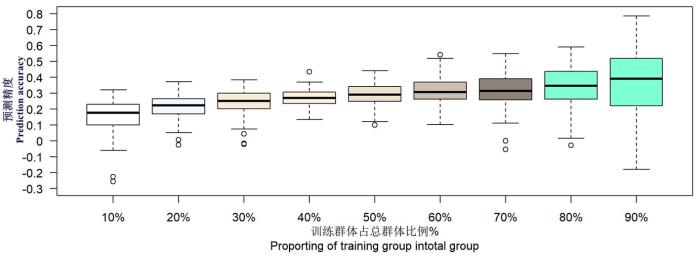
图2
F2群体不同训练群体大小玉米叶宽全基因组预测精度
Fig.2
Genomic selection accuracy of maize leaf width in different training populations in F2 population
当训练群体大小为总群体的10%时对叶宽进行全基因组选择分析,叶宽的预测精度在0.15上下均匀分布;当训练群体大小为20%时,叶宽的预测精度在0.21上下均匀分布;当训练群体大小为30%时,叶宽的预测精度均在0.24附近均匀分布;当训练群体大小为40%时,叶宽的预测精度均在0.27附近均匀分布;当训练群体大小上升到50%时,叶宽的预测精度在0.29附近均匀分布;之后当继续增大训练群体时,叶宽的预测精度始终在0.29附近均匀分布,变异范围逐渐增大。当训练群体大小为总群体的50%时进行全基因组选择。图3
图3
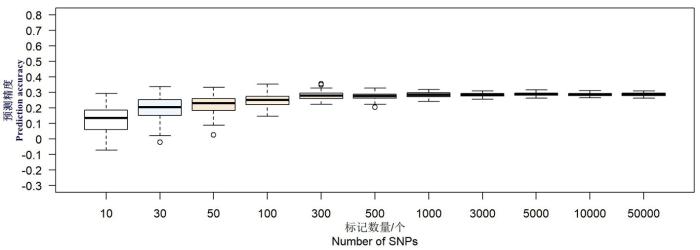
图3
F2群体不同SNP个数玉米叶宽全基因组预测精度
Fig.3
Genomic selection accuracy of leaf width with different SNP numbers in F2 population
3 讨论
研究在不同环境下共定位到12个与叶宽有关的QTL。其中qew5-1、qew4-1和qew5-2是主效QTL,表型贡献率分别为16.17%、10.63%和13.18%。其中qew1-1与qew5-1是多环境下稳定遗传的位点,分别位于1号和5号染色体。
唐登国[4]利用自交系B73和1212构建的325个重组自交系群体,采用完备区间作图法对叶宽进行QTL定位,定位到qLW1-5a和qLW1-5b定位区间为bin 5.01,与研究所定位qew5-1区间一致。文献[18]使用Yu82X9 D132组建RIL群体结合1 226个SNP标记在3种环境中进行QTL定位,定位到qLW8定位区间为bin 8.02和试验所定位到的qew8-1的定位区间一致。张莹莹等[5]在5号染色体与8号染色体上共定位到5个穗位叶QTL,其中在8号染色体中定位到的qSecLW1-8在bin 8.05定位区间内,和研究的qew8-2的定位区间相近。郑祖平等[19]用自交系Mo17与黄早四构建的RIL定位群体,定位到qWL4-1、qWL4-2和qWL10-1分别位于bin 4.08、bin 4.09和bin 10.07定位区间内,与试验qew4-1、qew4-2和qew10-1的定位区间一致。共定位到的QTL是稳定QTL,可以用于精细定位和基因克隆,研究定位结果的可靠。试验中,qew1-1属于微效新QTL位点,为叶宽遗传育种提供了新的变异位点。研究结果表明叶宽是由多个位点控制,以加性效应为主。
4 结论
4.1 玉米穗位叶宽受环境、基因型、环境与基因型互作显著影响。叶宽遗传力为0.39,遗传力较低,在育种时可以在较晚世代进行选择。
4.2 共检测到12个影响玉米穗位叶宽的QTL位点,分布在第1、3、4、5、8和10号染色体上,得到了2个多环境下稳定遗传的QTL位点,分别位于bin 1.06、bin 5.01,其中bin 5.01的QTL为主效位点,平均PVE为14.68%。
4.3 在双亲后代群体中,当训练群体比例占到群体基因型总数的50%、分子标记的个数到达300时,预测精度就可以到达相对较高的水平。
参考文献
Integrated multiple population analysis of leaf architecture traits in maize (Zea mays L.)
[J].
玉米穗三叶叶宽QTL定位及Meta分析
[J].
为了鉴定控制玉米穗三叶叶宽的主效QTL,以玉米自交系郑58和D863F为亲本,构建了包含241个家系的重组自交系群体,对河南原阳、西平以及海南乐东3个环境下的玉米穗三叶叶宽进行表型测定,利用215对SSR标记构建遗传图谱,对3个环境下的玉米穗三叶叶宽进行QTL定位研究。结果表明,郑58与D863F的穗三叶叶宽存在极显著差异,且RIL群体中穗三叶叶宽表现出连续变异,基本符合正态分布,属于典型的数量性状。3个环境下的穗三叶叶宽共定位到17个QTL,单个QTL解释表型变异率为5.30%~12.77%。其中,有3个QTL分别在2个及以上环境同时检测到,是控制穗三叶叶宽的主效QTL。通过Meta-QTL分析共得到12个mQTL,检测到的5号染色体上的qThiLW2-5位于mQTL5-1区段内,qFirLW2-6位于mQTL6-1区段内,qFirLW1-8、qFirLW2-8、qSecLW2-8位于mQTL8-1区段内。
QTL localization and Meta-analysis
[J].
玉米叶型相关性状QTL定位及上位性效应分析
[J].
QTL localization and epistatic effects analysis of Leaf-type-related traits in maize
[J].
全基因组选择模型研究进展及展望
[J].
Progress and outlook of genome-wide selection model
[J].
Molecular Markers and Selection for Complex Traits in Plants:Learning from the Last 20 Years
[J].
Genomic selection in hybrid breeding
[J].
不同水分环境下玉米叶形相关性状与SSR标记的关联分析
[J].
Association analysis of maize leaf shape-related traits and SSR markers in different moisture environments
[J].
花期玉米(Zea mays)穗三叶叶形结构的遗传效应及配合力分析
[J].
Genetic effect and coordination force analysis of the leaf shape structure of flowering maize (Zea mays)
[J].
Version 4.1 of QTL Ici Mapping:Integrated software for genetic linkage map construction and QTL mapping in bi-parental populations
[C]
Molecular-marker-facilitated investigations of quantitative-trait loci in maize.I.Numbers, genomic distribution and types of gene action
[J].Individual genetic factors which underlie variation in quantitative traits of maize were investigated in each of two F2 populations by examining the mean trait expressions of genotypic classes at each of 17-20 segregating marker loci. It was demonstrated that the trait expression of marker locus classes could be interpreted in terms of genetic behavior at linked quantitative trait loci (QTLs). For each of 82 traits evaluated, QTLs were detected and located to genomic sites. The numbers of detected factors varied according to trait, with the average trait significantly influenced by almost two-thirds of the marked genomic sites. Most of the detected associations between marker loci and quantitative traits were highly significant, and could have been detected with fewer than the 1800-1900 plants evaluated in each population. The cumulative, simple effects of marker-linked regions of the genome explained between 8 and 40% of the phenotypic variation for a subset of 25 traits evaluated. Single marker loci accounted for between 0.3% and 16% of the phenotypic variation of traits. Individual plant heterozygosity, as measured by marker loci, was significantly associated with variation in many traits. The apparent types of gene action at the QTLs varied both among traits and between loci for given traits, although overdominance appeared frequently, especially for yield-related traits. The prevalence of apparent overdominance may reflect the effects of multiple QTLs within individual marker-linked regions, a situation which would tend to result in overestimation of dominance. Digenic epistasis did not appear to be important in determining the expression of the quantitative traits evaluated. Examination of the effects of marked regions on the expression of pairs of traits suggests that genomic regions vary in the direction and magnitudes of their effects on trait correlations, perhaps providing a means of selecting to dissociate some correlated traits. Marker-facilitated investigations appear to provide a powerful means of examining aspects of the genetic control of quantitative traits. Modifications of the methods employed herein will allow examination of the stability of individual gene effects in varying genetic backgrounds and environments.
Epistatic and QTL×environment interaction effects on leaf area-associated traits in maize
[J].
不同供氮水平下玉米株型相关性状的QTLs定位和上位性效应分析
[J].
Localization of QTLs and epistatic effect analysis of type-related traits in maize at different nitrogen supply levels
[J].
Genome-Wide Analysis of Tar Spot Complex Resistance in Maize Using Genotyping-by-Sequencing SNPs and Whole-Genome Prediction
[J].
Genetic Dissection of Resistance to Common Rust (Puccinia Sorghi) in Tropical Maize (Zea mays L.) by Combined Genetic Mapping and Genomic Prediction
[J].

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}