金花菜与苜蓿属主要物种基因组SSR分布特征的比较分析
周勃 , 1 , 任海龙 1 , 2 , 张龑 3 , 高强 3 , 徐麟 3 , 邹集文 , 2
1.新疆农业科学院海南三亚农作物育种试验中心,海南三亚 572014
2.广州市农业科学研究院,广州 510308
3.新疆农业科学院农作物品种资源研究所,乌鲁木齐 830091
Characteristics and Analysis of Simple Sequence Repeats(SSR) in Medicago polymorpha and Main Medicagospecies genome
ZHOU Bo , 1 , REN Hailong 1 , 2 , ZHANG Yan 3 , GAO Qiang 3 , XU Lin 3 , ZOU Jiwen , 2
1. Hainan center of Xinjiang Academy of Agricultural Sciences, Sanya Hainan 572014, China
2. Guangzhou Institute of Agriculture Science, Guangzhou 510308, China
3. Institute of Crop Germplasm Resources, Xinjiang Academy of Agricultural Sciences, Urumqi 830091, China
通讯作者: 邹集文(1965-),男,广东梅州人,高级农艺师,研究方向为蔬菜种质资源保存与利用,(E-mail)13922740078@163.com
收稿日期: 2021-12-6
基金资助:
海南省自然科学基金 (319QN264 )广州市级财政补助资金农业农村项目 (20101292 )广州市支农资金项目 ([2021] 12 )
Corresponding authors: ZOU Jiwen (1965-), male, native place: Meizhou, Guangdong, native place: preservation and utilization of vegetable germplasm resources, (E-mail)13922740078@163.com
Received: 2021-12-6
Fund supported:
Natural Science Foundation of Hainan (319QN264 )Agricultural and Rural Project of Guangzhou Municipal Financial Subsidy Fund (20101292 )Agricultural Fund Project of Guangzhou ([2021] 12 )
摘要
【目的】 分析金花菜基因组SSR序列的分布特征,并与苜蓿属主要物种进行比较,为金花菜SSR分子标记的开发提供理论依据。【方法】 利用MISA软件对金花菜、蒺藜苜蓿和紫花苜蓿的高质量基因组进行搜索,比较分析搜索到的SSR序列分布特征。【结果】 在金花菜、蒺藜苜蓿和紫花苜蓿的基因组中,分别筛选到195 753个,242 434个和390 496个完整的SSR序列,相对密度分别为428、564和478个/Mb,SSR序列的总长度分别为3 611 698、3 657 503和6 307 211 bp,占各自基因组序列总长度的0.79%、0.85%和0.77%。在1~6个不同核苷酸重复单元中,金花菜和蒺藜苜蓿的SSR序列均是单核苷酸重复单元最多,依次是二核苷酸、三核苷酸、四核苷酸、五核苷酸和六核苷酸,而紫花苜蓿的六核苷酸重复单元多于五核苷酸重复单元。A、T、AT、TA、AG和TC是3种苜蓿共有的常见重复单元类型,金花菜基因组低片段长度的SSR比例高于蒺藜苜蓿和紫花苜蓿。【结论】 金花菜基因组SSR的分布密度低于蒺藜苜蓿和紫花苜蓿,重复单元类型较丰富,具有较大的多态性标记开发潜力。
关键词:
金花菜 蒺藜苜蓿 紫花苜蓿 基因组 SSR
Abstract
【Objective】 The distribution characteristics of SSR sequence of the Medicago polymorpha genome were analyzed and compared with the main species of Medicago, in order to provide theoretical basis for the development of SSR molecular markers in Medicago polymorpha. 【Method】 MISA software was used to search the high-quality genome of Medicago polymorpha, Medicago truncatula and Medicago sativa, and then the SSR sequence distribution characteristics were compared and analyzed. 【Result】 In the genome of Medicago polymorpha, Medicago truncatula and Medicago sativa, 195,753, 242,434 and 390,496 complete SSR sequences were screened, with relative densities of 428 /Mb, 564 /Mb and 478 /Mb, respectively. Total length of SSR sequences were 3,611,698 bp, 3,657,503 bp and 6,307,211 bp accounted for 0.79%, 0.85% and 0.77% of the total genome sequence length respectively. Among the 1~6 different nucleotide repeat units, the single nucleotide repeat units were the most in Medicago polymorpha and Medicago truncatula, followed by dinucleotide, trinucleotide, tetranucleotide, pentanucleotide and hexanucleotide repeat units, while the hexanucleotide repeat units were more than pentanucleotide repeat units in Medicago sativa. A, T, AT, TA, AG and TC were common repeat unit types among the three alfalfa species, and the proportion of SSR with low fragment length in Medicago polymorphagenome was higher than that in Medicago truncatula and Medicago sativa.【Conclusion】 The SSR distribution densityin the genome of Medicago polymorpha was lower than that of Medicago truncatula and Medicago sativa. SSR repeat unit types in Medicago polymorphagenomeare abundant and have great potential for developing polymorphic markers, which has important application value in the genetic diversity and molecular marker-assisted breeding of Medicago polymorpha .
Keywords:
Medicago polymorpha Medicago truncatula Medicago sativa genome SSR
本文引用格式
周勃, 任海龙, 张龑, 高强, 徐麟, 邹集文. 金花菜与苜蓿属主要物种基因组SSR分布特征的比较分析 [J]. 新疆农业科学, 2022, 59(9): 2217-2223 DOI:10.6048/j.issn.1001-4330.2022.09.016
ZHOU Bo, REN Hailong, ZHANG Yan, GAO Qiang, XU Lin, ZOU Jiwen. Characteristics and Analysis of Simple Sequence Repeats(SSR) in Medicago polymorpha and Main Medicagospecies genome [J]. Xinjiang Agricultural Sciences 10.6048/j.issn.1001-4330.2022.09.016
0 引言
【研究意义】金花菜(Medicago polymorpha )属豆科苜蓿属一年生苜蓿[1 ] 。金花菜在食用、饲用、药用和绿肥有较高价值[2 ] ,早年金花菜在我国栽培面积达20×104 hm2 (300万亩)[3 ] 。近年来,金花菜是我国极具发展前景的多用途豆科牧草[4 ] 。金花菜其所在的豆科苜蓿属(Medicago )大约有87个种,包括了豆科模式植物蒺藜苜蓿(Medicago truncatula )和最重要的豆科牧草紫花苜蓿(Medicago sativa )[5 ] 。金花菜作为苜蓿属“Polymorpha clade”进化分支的代表性物种,其染色体数目的非整倍体减少(基本染色体数8→7)[6 ] 。利用金花菜全基因组测序数据,分析其基因组中简单重复序列的分布特征及与蒺藜苜蓿和紫花苜蓿的异同,对金花菜种质资源遗传多样性和分子标记辅助选育有重要意义。【前人研究进展】简单重复序列(SSR, Simple Sequence Repeats)又称微卫星,为共显性标记,具有扩增稳定、数量丰富、多态性高及特异性强等优势[7 ] 、指纹图谱构建[8 ] 、遗传连锁图谱[9 ] 及QTLs定位等研究[10 ] 。利用高通量测序数据开发SSR标记是一种快速、高效、低成本的策略。由于金花菜等一年生苜蓿缺乏基因组序列信息,Eujayl等[11 ] 提出利用豆科模式植物蒺藜苜蓿的ESTs(Expressed sequence tags)序列,开发可用于其它一年生苜蓿的EST-SSR穿梭标记,89%的蒺藜苜蓿EST-SSRs在其他一年生苜蓿上可以跑出条带。Chu等[12 ] 通过对92对蒺藜苜蓿基因组SSR的研究发现,有53%的蒺藜苜蓿基因组SSR标记可以在金花菜上通用。但由于这些种间的SSR穿梭标记通常来自于物种基因组的保守区域,检测得到的金花菜多样性并不高,且难以获得大量的有效标记[13 ] 。【本研究切入点】由于缺少基因组信息,金花菜SSR标记的开发只能借鉴其近缘物种的基因组进行,制约着金花菜相关工作的有效开展。需找到均匀覆盖金花菜全基因组的分子标记并高通量开发。2021年金花菜的全基因组测序工作顺利完成,针对该物种基因组进行SSR标记的分析与开发成为可能。【拟解决的关键问题】在perl语言环境下,运行微卫星筛选软件MISA(MIcroSAtellite identification tool)的脚本,分别对金花菜、蒺藜苜蓿和紫花苜蓿的基因组FASTA文件进行扫描,对筛选出的简单重复序列进行统计分析。
1 材料与方法
1.1 材料
1.2 方法
使用微卫星检索工具MISA[17 ] (https://webblast.ipk-gatersleben.de/misa/ )执行命令perl misa.pl genome.fasta,对3种苜蓿全基因组进行扫描,筛选符合条件的简单重复序列。筛选标准为MISA软件的默认值:单核苷酸重复次数在10次及以上,二核苷酸重复次数在6次及以上,三至六核苷酸重复次数在5次及以上,复合型SSR的检索条件是2个SSR片段间的距离低于100 bp。将生成的数据采用Excel软件整理,对序列特征进行分析并绘制图表。
2 结果与分析
2.1 金花菜基因组SSR总体分布特征的比较
研究表明,金花菜为同源二倍体,染色体数目为14条,全基因组大小为457.53 Mb,共筛选出195 753个SSR,相对密度为428个/Mb,平均长度为18 bp;蒺藜苜蓿为同源二倍体,染色体数目为16条,全基因组大小为430.01 Mb,共筛选出242 434个SSR,相对密度为564个/Mb,平均长度为15 bp;紫花苜蓿为同源四倍体,染色体数目为32条,全基因组大小为817.12 Mb,共筛选出390 496个SSR,相对密度为478个/Mb,平均长度为16 bp。在这3种苜蓿中,金花菜检索到的SSR最少,平均SSR长度最长;蒺藜苜蓿检索到的SSR密度最高,平均SSR长度最短;紫花苜蓿的基因组最大,SSR的总数量最多。表2
2.2 金花菜基因组SSR不同核苷酸重复单元特征比较
研究表明,金花菜基因组SSR类型比较丰富,其中又以单核苷酸重复单元的数量最多,占基因组SSR数量的75.58%(147 953个SSR位点),其次为二、三核苷酸重复单元类型,分别占基因组SSR数量的15.31%(29 975个SSR位点)和7.94%(15 548个SSR位点);四、五、六核苷酸重复单元类型所占比例均相对较低,三者的比例总和仅为1.16%(共2 277个SSR位点)。金花菜和蒺藜苜蓿基因组中,SSR均是单核苷酸重复单元数目最多,然后依次是二核苷酸、三核苷酸、四核苷酸、五核苷酸和六核苷酸。紫花苜蓿除六核苷酸重复单元略高于五核苷酸重复单元外,其余核苷酸重复单元数目的变化趋势与金花菜和蒺藜苜蓿相一致,均是随重复单元核苷酸数的增加逐渐减少。
3种苜蓿同一核苷酸重复单元拷贝数变化趋势是相似的,且均随着重复拷贝数的增加,其SSR数目逐渐递减。金花菜,蒺藜苜蓿和紫花苜蓿的单核苷酸重复单元拷贝数主要集中在10~25次,分别占单核苷酸类型SSR总数的99.34%,98.95%和99.33%;二核苷酸重复单元拷贝数主要集中在6~26次,分别占比92.94%,91.20%和88.74%;三核苷酸重复单元拷贝数范围较为集中,主要集中在5~13次,分别占比94.21%,95.18%和88.81%;四核苷酸中重复单元拷贝数范围也较为集中,主要集中在5~8次,分别占比93.95%,96.57%和91.25%;五核苷酸重复单元拷贝数主要集中在5~7次,占比96.41%,98.03%和94.53%;六核苷酸重复单元拷贝数主要集中在5~7次,占比92.17%,94.53%和91.20%。在3种苜蓿中,金花菜的单核苷酸和二核苷酸重复单元拷贝数更为集中,蒺藜苜蓿三、四、五、六核苷酸重复单元拷贝数更为集中,紫花苜蓿的核苷酸重复单元拷贝数比金花菜和蒺藜苜蓿较为分散。表3
2.3 金花菜基因组SSR重复单元核苷酸构成的比较
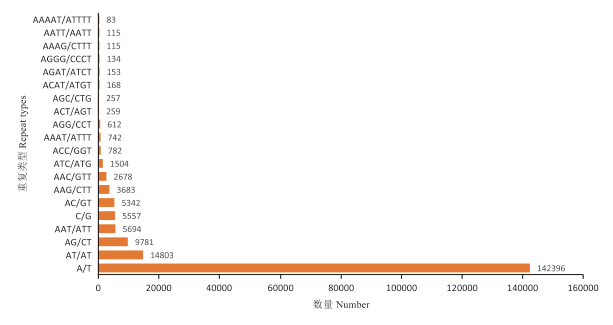
研究表明,共发现184种重复单元类型,比例最高的前20种类型共计有194 858个SSR(占99.54%)。在这些核苷酸重复单元中,单核苷酸重复单元中以A/T占绝对优势(共142 396个,占72.74%),其次为C/G(共5 557个,占2.84%);二核苷酸重复单元中以AT/AT占绝对优势(共14 803个,占7.56%),其次为AG/CT(共9 781个,占5.00%);三核苷酸重复类型中则以AAT/ATT(共5 694个,占2.91%)为优势重复单元类型,其次为AAG/CTT(1.88%)、AAC/GTT(1.37%)、ATC/ATG(0.77%)和ACC/GGT(0.40%)重复单元类型;四、五、六核苷酸重复单元相对较少,但类型较丰富,共2 277个,占SSR总数的1.16%,其中有72种重复单元类型仅出现1次。图1
图 1
图 1
金花菜基因组SSR的重复基元类型及数量
Fig. 1
The number of different SSRin Medicago polymorpha genome
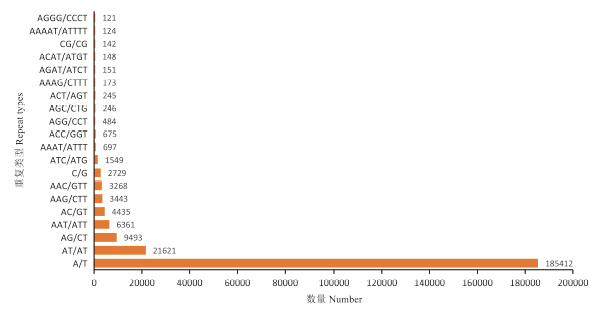
蒺藜苜蓿基因组中共发现177种重复单元类型,比例最高的20种类型SSR共计241 517个(占99.62%),从高到低依次为:A/T(共185 412个,占76.48%)、AT/AT(共21 621个,占8.92%)、AG/CT(共9 493个,占3.92%)、AAT/ATT(共6 361个,占2.62%)、AC/GT(共4 435个,占1.83%)、AAG/CTT(共3 443个,占1.42%)、AAC/GTT(共3 268个,占1.35%)、C/G(共2 729个,占1.13%)、ATC/ATG(共1 549个,占0.64%)、AAAT/ATTT(共697个,占0.29%)、ACC/GGT(共675个,占0.28%)、AGG/CCT(共484个,占0.20%)、AGC/CTG(共246个,占0.10%)、ACT/AGT(共245个,占0.10%)、AAAG/CTTT(共173个,占0.07%)、AGAT/ATCT(共151个,占0.06%)、ACAT/ATGT(共148个,占0.06%)、CG/CG(共142个,占0.06%)、AAAAT/ATTTT(共124个,占0.05%)和AGGG/CCCT(共121个,占0.05%)。图2
图 2
图 2
蒺藜苜蓿基因组SSR的重复基元类型及数量
Fig. 2
The number of different SSRin Medicago truncatula genome
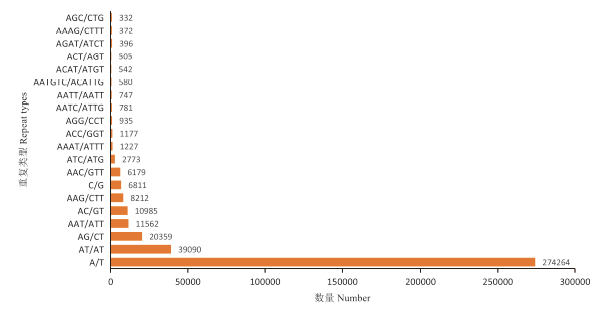
紫花苜蓿基因组中共发现200种重复单元类型,比例最高的20种类型SSR共计387 829个(占99.32%),从高到低依次为:A/T(共274 264个,占70.23%)、AT/AT(共39 090个,占10.01%)、AG/CT(共20 359个,占5.21%)、AAT/ATT(共11 562个,占2.96%)、AC/GT(共10 985个,占2.81%)、AAG/CTT(共8 212个,占2.10%)、C/G(共6 811个,占1.74%)、AAC/GTT(共6 179个,占1.58%)、ATC/ATG(共2 773个,占0.71%)、AAAT/ATTT(共1 227个,占0.31%)、ACC/GGT(共1 177个,占0.30%)、AGG/CCT(共935个,占0.24%)、AATC/ATTG(共781个,占0.20%)、AATT/AATT(共747个,占0.19%)、AATGTC/ACATTG(共580个,占0.15%)、ACAT/ATGT(共542个,占0.14%)、ACT/AGT(共505个,占0.13%)、AGAT/ATCT(共396个,占0.10%)、AAAG/CTTT(共372个,占0.10%)和AGC/CTG(共332个,占0.09%)。
3种苜蓿中,紫花苜蓿基因组中SSR重复单元类型最多,其次是金花菜和蒺藜苜蓿。重复单元类型中,A/T、AT/AT、AG/CT和AAT/ATT是三种苜蓿共有的常见核心SSR类型。图3
图 3
图 3
紫花苜蓿基因组SSR的重复基元类型及数量
Fig. 3
The number of different SSRin Medicago sativa genome
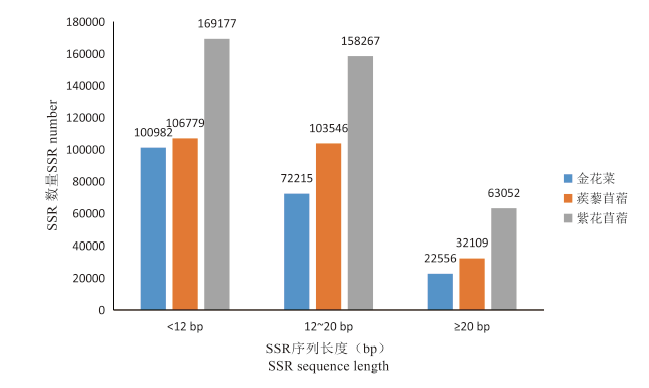
2.4 金花菜基因组SSR片段长度比较
研究表明,金花菜基因组SSR序列长度主要集中于12 bp以内,为100 982个,占SSR总数的51.59%;分布于12~20 bp的SSR数量为72 215个,占SSR总数的36.89%;≥20 bp以上的SSR数量为22 556个,占SSR总数的11.52%。相较于蒺藜苜蓿和紫花苜蓿,金花菜SSR分布在<12 bp长度上的比例最高。金花菜仍有大量的中等多态性和较高多态性长度的SSR,具有较大的多态性标记开发潜力。图4
图 4
图 4
金花菜基因组SSR不同片段长度的数量
Fig. 4
The number of different fragment lengthSSR in Medicago polymorpha genome
3 讨论
SSR序列长度<12 bp时SSR标记的多态性表现极低;序列长度在12~20 bp之间时标记多态性适中;≥20 bp时具有较高多态性,是理想的标记位点[18 ] 。基因组中存在着大量的重复序列,从进化角度看,物种间重复序列的差异是自然选择的结果,因此鉴定SSR在基因组中的分布特征有重要意义[19 ] 。金花菜、蒺藜苜蓿和紫花苜蓿是苜蓿属的不同种,其中金花菜和蒺藜苜蓿属于一年生苜蓿,紫花苜蓿属于多年生苜蓿,3种苜蓿基因组有很强的的共线性关系[14 ] 。研究发现,金花菜基因组SSR的分布密度为428个/Mb,明显低于蒺藜苜蓿的分布密度(564个/Mb)以及紫花苜蓿的分布密度(478个/Mb)。Varshney等[20 ] 研究认为,SSR分布密度之所以出现差异,除了物种间差异因素外,还与测序数据深度、序列拼接数据质量及SSR位点查找软件以及SSR搜索标准不同有关。研究选用主流的微卫星筛选软件MISA(MIcroSAtellite identification tool),在相同设置条件下分析了这3种苜蓿间差异。金花菜、蒺藜苜蓿和紫花苜蓿的测序深度分别为117X、109X和153X,均为二代+三代测序组装的高质量基因组,结果比较能真实发映出物种间的差异。金花菜基因组SSR的分布密度较低,可能与金花菜染色体数目少有关。
单核苷酸、二核苷酸和三核苷酸重复单元是绝大多植物基因组SSR序列中优势重复单元[18 ,21 ] 。研究发现,金花菜基因组SSR中,单、二和三核苷酸重复单元类型分别占基因组SSR数量的75.58%、15.31%和7.94%,其后依次是,四、五和六核苷酸重复单元,与蒺藜苜蓿观测到的结果相一致。与金花菜和蒺藜苜蓿相比,紫花苜蓿的六核苷酸重复单元数量多于五核苷酸重复单元,且单核苷酸重复单元类型的SSR数量相对较少,这可能与紫花苜蓿是同源四倍体,异花授粉导致遗传变异更为丰富有关。
4 结论
共发现94 771个片段长度≥12 bp的金花菜基因组简单重复序列,具有较高的多态性。
金花菜基因组共筛选出195 753个SSR,相对密度为428个/Mb,平均长度为18 bp,金花菜基因组SSR的分布密度低于蒺藜苜蓿和紫花苜蓿,重复单元类型较丰富,具有较大的多态性标记开发潜力。
参考文献
View Option
[1]
任海龙 , 魏臻武 , 陈祥 . 金花菜应用研究进展
[J]. 中国野生植物资源 , 2014 , 33 (5 ):33 -36 .
[本文引用: 1]
REN Hailong WEI Zhenwu CHEN Xiang Development and application of burclover
[J]. Chinese Wild Plant Resources 2014 , 33 (5 ): 33 -36 .
[本文引用: 1]
[2]
侯祥川 , 李建新 . 富有营养的野菜苜蓿
[J]. 人民军医 , 1951 ,(5 ):5 -7 .
[本文引用: 1]
HOU Xiangchuan LI Jianxin Nutritious wild clover
[J]. People's Military Surgeon 1951 , (5 ): 5 -7 .
[本文引用: 1]
[3]
陈一吾 , 吴仁润 . 华中和华南的苜蓿与南苜蓿的生产
[J]. 草与畜杂志 , 1987 ,(6 ):30 -31 .
[本文引用: 1]
CHEN Yiwu WU Renrun The production of alfalfaandburcloverinCentralChinaandSouthernChina
[J]. China Herbivore Science 1987 , (6 ): 30 -31 .
[本文引用: 1]
[4]
曹德明 , 魏臻武 , 虞珍萍 , 等 . 扬中金花菜产业发展新模式——南方草业的新亮点
[J]. 草原与草坪 , 2012 , 32 (5 ):79 -82 .
[本文引用: 1]
CAO Deming WEI Zhenwu YU Zhenping et al. Newmodelof tootedburcloverindustryinYangzhong city—anewbright spotofprataculturedevelopment of China
[J]. Grass land and Turf 2012 , 32 (5 ): 79 -82 .
[本文引用: 1]
[5]
Steele KP Ickert-Bond SM Zarre S et al. Phylogeny and character evolution in Medicago (Leguminosae): Evidence from analyses of plastid trnK/matK and nuclear GA3ox1 sequences
[J]. American Journal of Botany 2010 , 97 : 1142 -1155 .
DOI
PMID
[本文引用: 1]
The genus Medicago, with about 87 species, includes the model legume species M. truncatula, and a number of important forage species such as M. sativa (alfalfa), M. scutellata (snail medic), and M. lupulina (black medic). Relationships within the genus are not yet sufficiently resolved, contributing to difficulty in understanding the evolution of a number of distinguishing characteristics such as aneuploidy and polyploidy, life history, structure of cotyledons, and number of seeds per fruit. •Phylogenetic relationships of 70-73 species of Medicago and its sister genus Trigonella (including Melilotus) were reconstructed from nucleotide sequences of the plastid trnK/matK region and the nuclear-encoded GA3ox1 gene (gibberellin 3-β-hydroxylase) using maximum parsimony and Bayesian inference methods. •Our results support certain currently recognized taxonomic groups, e.g., sect. Medicago (with M. sativa) and sect. Buceras. However, other strongly supported clades-the "reduced subsection Leptospireae clade" that includes M. lupulina, the "polymorpha clade" that includes M. murex and M. polymorpha and the "subsection Pachyspireae clade" that includes M. truncatula-each of which includes species presently in different subsections of sect. Spirocarpos, contradict the current classification. •These results support the hypothesis that some characters considered important in existing taxonomies, for example, single-seeded fruits that have arisen more than once in both Medicago and Trigonella, are indeed homoplastic. Others, such as the 2n = 14 chromosome number, have also arisen independently within the genus. In addition, we demonstrate support for the utility of GA3ox1 sequences for phylogenetic analysis among and within closely related genera of legumes.
[6]
Yoder JB Briskine R Mudge J et al. Phylogenetic signal variation in the genomes of Medicago (fabaceae)
[J]. Systematic Biology 2013 , 62 (3 ): 424 -438 .
DOI
URL
[本文引用: 1]
[7]
麻冬梅 , 张喜斌 , 黄婷 , 等 . 利用SSR标记对不同耐盐紫花苜蓿遗传多样性分析
[J]. 草地学报 , 2019 , 27 (6 ):1477 -1485 .
DOI
[本文引用: 1]
为了了解不同耐盐紫花苜蓿(Medicago sativa L.)的遗传多样性以及对耐盐紫花苜蓿品种改良,本试验筛选出9对简单重复序列(Simple Sequence Repeats,SSR)引物对10份耐盐、敏盐材料进行SSR多态性分析。并利用MEGA6.0软件对其进行了聚类分析。结果表明:10份紫花苜蓿品种中平均多态位点百分率为47.1%~71.8%;平均有效等位基因观察数与等位基因观察数的比为0.680~0.890;平均杂合度为0.759~0.828;平均香农指数为1.753~1.976;平均多态信息含量为0.735~0.811;遗传分化系数为0.100。利用IBM SPSS Statistics 19软件进行综合评价,根据综合评价值对10个紫花苜蓿品种内的遗传变异程度大小排序,排列顺序为: "巨能" > "骑士T" > "巨能7号" > "Asi" > "兴平" > "阿迪娜" > "秘鲁" > "抗旱15" > "草原3号" > "国产苜蓿";利用MEGA6.0软件对Nei遗传距离进行UPGMA聚类分析可知:5份敏盐材料始终聚为一类;"巨能"与"巨能7号"聚为一类;"阿迪娜"和"抗旱15"又聚为一类,说明他们之间有较近的亲缘关系。
MA Dongmei ZAHNG Xibin HUANG Ting et al. Genetic diversity analysis of 10 salt tolerant alfalfa
[J]. ActaAgrestia Sinica 2019 , 27 (6 ): 1477 -1485 .
[本文引用: 1]
[8]
张迪 , 任立飞 , 张璐璐 , 等 . 基于荧光毛细管电泳技术的苜蓿SSR指纹图谱构建
[J]. 中国草地学报 , 2020 , 42 (6 ):10 -14 .
[本文引用: 1]
ZHANG Di REN Lifei ZHANG Lulu et al. Construction of SSR fingerprint of Alfalfa based on fluorescent capillary electrophoresis
[J]. Chinese Journal of Grassland 2020 , 42 (6 ):10 -14 .
[本文引用: 1]
[9]
王梦颖 , 张铁军 , 龙瑞才 , 等 . 四倍体紫花苜蓿遗传图谱的初步构建
[J]. 草地学报 , 2015 , 23 (6 ):1247 -1251 .
DOI
[本文引用: 1]
分别以早熟低产和晚熟高产苜蓿单株为父母本,通过人工杂交构建了四倍体紫花苜蓿(Medicago Sativa)F1遗传作图群体,采用单因子变量分析法,以降落式PCR和常规PCR结合的反应程序,建立了适宜于紫花苜蓿的分子标记扩增体系;应用130对SSR引物进行筛选,获得60对引物在父母本间存在多态性而被用于绘制遗传连锁图。采用PAGE电泳分析,对作图群体进行基因型分析。通过TetraploidMap软件对60个SSR标记进行连锁作图分析,有44个标记可以定位在8个连锁群上,占总标记数的33.8%,覆盖遗传距离979 cM,两标记间平均图距为22.25 cM,初步构建了四倍体紫花苜蓿遗传图谱的框架图,还需要进一步添加标记数量增大其饱和度,为重要性状的QTL定位奠定基础。
WANG Mengying ZHANG Tiejun LONG Ruicai et al. Preliminary construction of genetic map in tet rap lo id alfalfa
Acta Agrestia Sinica 2015 , 23 (6 ): 1247 -1251 .
[本文引用: 1]
[10]
张世超 , 王英哲 , 金艳 , 等 . 紫花苜蓿细胞质雄性不育恢复基因的初步定位
[J]. 草业科学 , 2018 , 35 (5 ):1067 -1071 .
[本文引用: 1]
ZHANG Shichao WANG Yingzhe JIN Yan et al. The preliminary location study on the fertility restore genes for cytoplasmic Male sterility (CMS) inalfalfa
[J]. PrataculturalScience 2018 , 35 (5 ): 1067 -1071 .
[本文引用: 1]
[11]
Eujayl I Sledge MK Wang L et al. Medicago truncatula EST-SSRs reveal cross-species genetic markers for Medicago spp
[J]. Theoretical and Applied Genetics 2004 , 108 (3 ):414 -422 .
PMID
[本文引用: 1]
Expressed sequence tags (ESTs) are important resources for gene discovery and molecular marker development. From over 147,000 ESTs of Medicago truncatula, we have identified 4,384 ESTs containing perfect simple sequence repeats (EST-SSR) of di-, tri-, tetra- or pentanucleotides. Six hundred sixteen primer pairs (PPs) were designed and screened over a panel of eight genotypes representing six Medicago spp. and subspecies. Nearly, 74% (455) of the PPs produced characteristic SSR bands of expected size length in at least one Medicago species. Four hundred six (89%) of these 455 PPs produced SSR bands in all eight genotypes tested. Only 17 PPs were M. truncatula -specific. High levels of polymorphism (>70%) were detected for these markers in alfalfa, M. truncatula, and other annual medics. About 48% of the reported markers are part of gene transcripts linked to putative functions. Our results indicate that the SSR markers developed from M. truncatula ESTs are valuable genetic markers for the Medicago genus. These markers will be useful in establishing the genomic relationships of M. truncatula to important forage legume crops such as alfalfa and other annual medics.
[12]
Chu HJ Yan J Hu Y et al. Cross-species amplification of 92 microsatellites of Medicago truncatula
[J]. Molecular Ecology Resources 2010 , 10 : 150 -155 .
DOI
PMID
[本文引用: 1]
Medicago species are important genetic sources for forage crops and nitrogen sources for various ecosystems. The ongoing genome sequencing of the model legume, Medicago truncatula, provides a wealth of genetic markers potentially useful for characterizing the population genetic structure and evolutionary history, and the potential of the wild Medicago species. Here we tested the PCR amplification of 92 microsatellites developed from M. truncatula in six other Medicago species, and found that the cross-species transferability, ranging from 53.26% to 61.96%, is comparable with those reported in other angiosperm genera. This article thus reports a number of microsatellites that are potentially useful for large-scale ecological and evolutionary genetic studies of wild Medicago species.© 2009 Blackwell Publishing Ltd.
[13]
陈祥 , 魏臻武 , 任海龙 , 等 . 聚丙烯酰胺凝胶电泳检测南苜蓿SSR标记
[J]. 江苏农业科学 , 2015 , 43 (11 ):382 -384 .
[本文引用: 1]
CHEN Xiang WEI Zhenwu REN Hailong et al. Detection of SSR markers in burclover by polyacrylamide gel electrophoresis
[J]. Jiangsu Agricultural Sciences 2015 , 43 (11 ): 382 -384 .
[本文引用: 1]
[14]
Cui J Lu Z Wang T et al. The genome of Medicago polymorpha provides insights into its edibility and nutritional value as a vegetable and forage legume
[J]. Hortic Res 2021 , 8 (1 ), 47.
[本文引用: 2]
[15]
Pecrix Y Staton SE Sallet E et al. Whole-genome landscape of Medicago truncatula symbiotic genes
[J]. Nature Plants 2018 , 4 (12 ):1017 -1025 .
DOI
PMID
[本文引用: 1]
Advances in deciphering the functional architecture of eukaryotic genomes have been facilitated by recent breakthroughs in sequencing technologies, enabling a more comprehensive representation of genes and repeat elements in genome sequence assemblies, as well as more sensitive and tissue-specific analyses of gene expression. Here we show that PacBio sequencing has led to a substantially improved genome assembly of Medicago truncatula A17, a legume model species notable for endosymbiosis studies, and has enabled the identification of genome rearrangements between genotypes at a near-base-pair resolution. Annotation of the new M. truncatula genome sequence has allowed for a thorough analysis of transposable elements and their dynamics, as well as the identification of new players involved in symbiotic nodule development, in particular 1,037 upregulated long non-coding RNAs (lncRNAs). We have also discovered that a substantial proportion (~35% and 38%, respectively) of the genes upregulated in nodules or expressed in the nodule differentiation zone colocalize in genomic clusters (270 and 211, respectively), here termed symbiotic islands. These islands contain numerous expressed lncRNA genes and display differentially both DNA methylation and histone marks. Epigenetic regulations and lncRNAs are therefore attractive candidate elements for the orchestration of symbiotic gene expression in the M. truncatula genome.
[16]
Shen C Du H Chen Z et al. The Chromosome-Level Genome Sequence of the Autotetraploid Alfalfa and Resequencing of Core Germplasms Provide Genomic Resources for Alfalfa Research
[J]. Mol Plant 2020 , 13 (9 ):1250 -1261 .
DOI
PMID
[本文引用: 1]
Alfalfa (Medicago sativa) is one of the most important forage crops in the world; however, its molecular genetics and breeding research are hindered due to the lack of a high-quality reference genome. Here, we report a de novo assembled 816-Mb high-quality, chromosome-level haploid genome sequence for 'Zhongmu No.1' alfalfa, a heterozygous autotetraploid. The contig N50 is 3.92 Mb, and 49 165 genes are annotated in the genome. The alfalfa genome is estimated to have diverged from M. truncatula approximately 8 million years ago. Genomic population analysis of 162 alfalfa accessions revealed high genetic diversity, weak population structure, and extensive gene flow from wild to cultivated alfalfa. Genome-wide association studies identified many candidate genes associated with important agronomic traits. Furthermore, we showed that MsFTa2, a Flowering Locus T homolog, whose expression is upregulated in salt-resistant germplasms, may be associated with fall dormancy and salt resistance. Taken together, these genomic resources will facilitate alfalfa genetic research and agronomic improvement.Copyright © 2020 The Author. Published by Elsevier Inc. All rights reserved.
[17]
Beier S Thiel T Münch T, etal MISA-web: a web server for microsatellite prediction
[J]. Bioinformatics 2017 , 33 : 2583 -2585 .
DOI
PMID
[本文引用: 1]
Microsatellites are a widely-used marker system in plant genetics and forensics. The development of reliable microsatellite markers from resequencing data is challenging.We extended MISA, a computational tool assisting the development of microsatellite markers, and reimplemented it as a web-based application. We improved compound microsatellite detection and added the possibility to display and export MISA results in GFF3 format for downstream analysis.MISA-web can be accessed under http://misaweb.ipk-gatersleben.de/. The website provides tutorials, usage note as well as download links to the source code.scholz@ipk-gatersleben.de.© The Author(s) 2017. Published by Oxford University Press.
[18]
邱炳发 , 梁馨元 , 王建忠 , 等 . 大花序桉基因组SSR的分布特征及序列分析
[J]. 南方农业学报 , 2021 , 52 (10 ):2744 -2750 .
[本文引用: 2]
QIU Bingfa LIANG Xinyuan WANG Jianzhong et al. Characteristics and analysis of simple sequence repeats(SSR)in Eucalyptus cloeziana genome
[J]. Journal of Southern Agriculture 2021 , 52 (10 ): 2744 -2750 .
[本文引用: 2]
[19]
李新玉 , 王希胤 . 重复序列对植物基因组大小进化的影响
[J]. 华北理工大学学报(自然科学版) , 2021 , 43 (4 ):98 -107 .
[本文引用: 1]
LI Xinyu WANG Xiyin Effects of repetitive sequences to evolution of plant genome size
[J]. Journal of North China University of Science and Technology (NaturalScienceEdition )2021 , 43 (4 ): 98 -107 .
[本文引用: 1]
[20]
Varshney RK Graner A Sorrells ME Genic microsatellite markers in plants: Features and applications
[J]. Trends in Biotechnology 2005 , 23 (1 ): 48 -55 .
DOI
PMID
[本文引用: 1]
Expressed sequence tag (EST) projects have generated a vast amount of publicly available sequence data from plant species; these data can be mined for simple sequence repeats (SSRs). These SSRs are useful as molecular markers because their development is inexpensive, they represent transcribed genes and a putative function can often be deduced by a homology search. Because they are derived from transcripts, they are useful for assaying the functional diversity in natural populations or germplasm collections. These markers are valuable because of their higher level of transferability to related species, and they can often be used as anchor markers for comparative mapping and evolutionary studies. They have been developed and mapped in several crop species and could prove useful for marker-assisted selection, especially when the markers reside in the genes responsible for a phenotypic trait. Applications and potential uses of EST-SSRs in plant genetics and breeding are discussed.
[21]
Biet E Sun J Dutreix M Conserved sequence preference in DNA binding among recombination proteins: An effect of ssDNA secondary structure
[J]. Nucleic Acids Research 1999 , 27 (2 ): 596 -600 .
PMID
[本文引用: 1]
Repetitive sequences have been proposed to be recombinogenic elements in eukaryotic chromosomes. We tested whether dinucleotide repeats sequences are preferential sites for recombination because of their high affinity for recombination enzymes. We compared the kinetics of the binding of the scRad51, hsRad51 and ecRecA proteins to oligonucleotides with repeats of dinucleotides GT, CA, CT, GA, GC or AT. Since secondary structures in single-stranded DNA (ssDNA) act as a barrier to complete binding we measured whether these oligonucleotides are able to form stable secondary structures. We show that the preferential binding of recombination proteins is conserved among the three proteins and is influenced mainly by secondary structures in ssDNA.
金花菜应用研究进展
1
2014
... 【研究意义】金花菜(Medicago polymorpha )属豆科苜蓿属一年生苜蓿[1 ] .金花菜在食用、饲用、药用和绿肥有较高价值[2 ] ,早年金花菜在我国栽培面积达20×104 hm2 (300万亩)[3 ] .近年来,金花菜是我国极具发展前景的多用途豆科牧草[4 ] .金花菜其所在的豆科苜蓿属(Medicago )大约有87个种,包括了豆科模式植物蒺藜苜蓿(Medicago truncatula )和最重要的豆科牧草紫花苜蓿(Medicago sativa )[5 ] .金花菜作为苜蓿属“Polymorpha clade”进化分支的代表性物种,其染色体数目的非整倍体减少(基本染色体数8→7)[6 ] .利用金花菜全基因组测序数据,分析其基因组中简单重复序列的分布特征及与蒺藜苜蓿和紫花苜蓿的异同,对金花菜种质资源遗传多样性和分子标记辅助选育有重要意义.【前人研究进展】简单重复序列(SSR, Simple Sequence Repeats)又称微卫星,为共显性标记,具有扩增稳定、数量丰富、多态性高及特异性强等优势[7 ] 、指纹图谱构建[8 ] 、遗传连锁图谱[9 ] 及QTLs定位等研究[10 ] .利用高通量测序数据开发SSR标记是一种快速、高效、低成本的策略.由于金花菜等一年生苜蓿缺乏基因组序列信息,Eujayl等[11 ] 提出利用豆科模式植物蒺藜苜蓿的ESTs(Expressed sequence tags)序列,开发可用于其它一年生苜蓿的EST-SSR穿梭标记,89%的蒺藜苜蓿EST-SSRs在其他一年生苜蓿上可以跑出条带.Chu等[12 ] 通过对92对蒺藜苜蓿基因组SSR的研究发现,有53%的蒺藜苜蓿基因组SSR标记可以在金花菜上通用.但由于这些种间的SSR穿梭标记通常来自于物种基因组的保守区域,检测得到的金花菜多样性并不高,且难以获得大量的有效标记[13 ] .【本研究切入点】由于缺少基因组信息,金花菜SSR标记的开发只能借鉴其近缘物种的基因组进行,制约着金花菜相关工作的有效开展.需找到均匀覆盖金花菜全基因组的分子标记并高通量开发.2021年金花菜的全基因组测序工作顺利完成,针对该物种基因组进行SSR标记的分析与开发成为可能.【拟解决的关键问题】在perl语言环境下,运行微卫星筛选软件MISA(MIcroSAtellite identification tool)的脚本,分别对金花菜、蒺藜苜蓿和紫花苜蓿的基因组FASTA文件进行扫描,对筛选出的简单重复序列进行统计分析. ...
金花菜应用研究进展
1
2014
... 【研究意义】金花菜(Medicago polymorpha )属豆科苜蓿属一年生苜蓿[1 ] .金花菜在食用、饲用、药用和绿肥有较高价值[2 ] ,早年金花菜在我国栽培面积达20×104 hm2 (300万亩)[3 ] .近年来,金花菜是我国极具发展前景的多用途豆科牧草[4 ] .金花菜其所在的豆科苜蓿属(Medicago )大约有87个种,包括了豆科模式植物蒺藜苜蓿(Medicago truncatula )和最重要的豆科牧草紫花苜蓿(Medicago sativa )[5 ] .金花菜作为苜蓿属“Polymorpha clade”进化分支的代表性物种,其染色体数目的非整倍体减少(基本染色体数8→7)[6 ] .利用金花菜全基因组测序数据,分析其基因组中简单重复序列的分布特征及与蒺藜苜蓿和紫花苜蓿的异同,对金花菜种质资源遗传多样性和分子标记辅助选育有重要意义.【前人研究进展】简单重复序列(SSR, Simple Sequence Repeats)又称微卫星,为共显性标记,具有扩增稳定、数量丰富、多态性高及特异性强等优势[7 ] 、指纹图谱构建[8 ] 、遗传连锁图谱[9 ] 及QTLs定位等研究[10 ] .利用高通量测序数据开发SSR标记是一种快速、高效、低成本的策略.由于金花菜等一年生苜蓿缺乏基因组序列信息,Eujayl等[11 ] 提出利用豆科模式植物蒺藜苜蓿的ESTs(Expressed sequence tags)序列,开发可用于其它一年生苜蓿的EST-SSR穿梭标记,89%的蒺藜苜蓿EST-SSRs在其他一年生苜蓿上可以跑出条带.Chu等[12 ] 通过对92对蒺藜苜蓿基因组SSR的研究发现,有53%的蒺藜苜蓿基因组SSR标记可以在金花菜上通用.但由于这些种间的SSR穿梭标记通常来自于物种基因组的保守区域,检测得到的金花菜多样性并不高,且难以获得大量的有效标记[13 ] .【本研究切入点】由于缺少基因组信息,金花菜SSR标记的开发只能借鉴其近缘物种的基因组进行,制约着金花菜相关工作的有效开展.需找到均匀覆盖金花菜全基因组的分子标记并高通量开发.2021年金花菜的全基因组测序工作顺利完成,针对该物种基因组进行SSR标记的分析与开发成为可能.【拟解决的关键问题】在perl语言环境下,运行微卫星筛选软件MISA(MIcroSAtellite identification tool)的脚本,分别对金花菜、蒺藜苜蓿和紫花苜蓿的基因组FASTA文件进行扫描,对筛选出的简单重复序列进行统计分析. ...
富有营养的野菜苜蓿
1
1951
... 【研究意义】金花菜(Medicago polymorpha )属豆科苜蓿属一年生苜蓿[1 ] .金花菜在食用、饲用、药用和绿肥有较高价值[2 ] ,早年金花菜在我国栽培面积达20×104 hm2 (300万亩)[3 ] .近年来,金花菜是我国极具发展前景的多用途豆科牧草[4 ] .金花菜其所在的豆科苜蓿属(Medicago )大约有87个种,包括了豆科模式植物蒺藜苜蓿(Medicago truncatula )和最重要的豆科牧草紫花苜蓿(Medicago sativa )[5 ] .金花菜作为苜蓿属“Polymorpha clade”进化分支的代表性物种,其染色体数目的非整倍体减少(基本染色体数8→7)[6 ] .利用金花菜全基因组测序数据,分析其基因组中简单重复序列的分布特征及与蒺藜苜蓿和紫花苜蓿的异同,对金花菜种质资源遗传多样性和分子标记辅助选育有重要意义.【前人研究进展】简单重复序列(SSR, Simple Sequence Repeats)又称微卫星,为共显性标记,具有扩增稳定、数量丰富、多态性高及特异性强等优势[7 ] 、指纹图谱构建[8 ] 、遗传连锁图谱[9 ] 及QTLs定位等研究[10 ] .利用高通量测序数据开发SSR标记是一种快速、高效、低成本的策略.由于金花菜等一年生苜蓿缺乏基因组序列信息,Eujayl等[11 ] 提出利用豆科模式植物蒺藜苜蓿的ESTs(Expressed sequence tags)序列,开发可用于其它一年生苜蓿的EST-SSR穿梭标记,89%的蒺藜苜蓿EST-SSRs在其他一年生苜蓿上可以跑出条带.Chu等[12 ] 通过对92对蒺藜苜蓿基因组SSR的研究发现,有53%的蒺藜苜蓿基因组SSR标记可以在金花菜上通用.但由于这些种间的SSR穿梭标记通常来自于物种基因组的保守区域,检测得到的金花菜多样性并不高,且难以获得大量的有效标记[13 ] .【本研究切入点】由于缺少基因组信息,金花菜SSR标记的开发只能借鉴其近缘物种的基因组进行,制约着金花菜相关工作的有效开展.需找到均匀覆盖金花菜全基因组的分子标记并高通量开发.2021年金花菜的全基因组测序工作顺利完成,针对该物种基因组进行SSR标记的分析与开发成为可能.【拟解决的关键问题】在perl语言环境下,运行微卫星筛选软件MISA(MIcroSAtellite identification tool)的脚本,分别对金花菜、蒺藜苜蓿和紫花苜蓿的基因组FASTA文件进行扫描,对筛选出的简单重复序列进行统计分析. ...
富有营养的野菜苜蓿
1
1951
... 【研究意义】金花菜(Medicago polymorpha )属豆科苜蓿属一年生苜蓿[1 ] .金花菜在食用、饲用、药用和绿肥有较高价值[2 ] ,早年金花菜在我国栽培面积达20×104 hm2 (300万亩)[3 ] .近年来,金花菜是我国极具发展前景的多用途豆科牧草[4 ] .金花菜其所在的豆科苜蓿属(Medicago )大约有87个种,包括了豆科模式植物蒺藜苜蓿(Medicago truncatula )和最重要的豆科牧草紫花苜蓿(Medicago sativa )[5 ] .金花菜作为苜蓿属“Polymorpha clade”进化分支的代表性物种,其染色体数目的非整倍体减少(基本染色体数8→7)[6 ] .利用金花菜全基因组测序数据,分析其基因组中简单重复序列的分布特征及与蒺藜苜蓿和紫花苜蓿的异同,对金花菜种质资源遗传多样性和分子标记辅助选育有重要意义.【前人研究进展】简单重复序列(SSR, Simple Sequence Repeats)又称微卫星,为共显性标记,具有扩增稳定、数量丰富、多态性高及特异性强等优势[7 ] 、指纹图谱构建[8 ] 、遗传连锁图谱[9 ] 及QTLs定位等研究[10 ] .利用高通量测序数据开发SSR标记是一种快速、高效、低成本的策略.由于金花菜等一年生苜蓿缺乏基因组序列信息,Eujayl等[11 ] 提出利用豆科模式植物蒺藜苜蓿的ESTs(Expressed sequence tags)序列,开发可用于其它一年生苜蓿的EST-SSR穿梭标记,89%的蒺藜苜蓿EST-SSRs在其他一年生苜蓿上可以跑出条带.Chu等[12 ] 通过对92对蒺藜苜蓿基因组SSR的研究发现,有53%的蒺藜苜蓿基因组SSR标记可以在金花菜上通用.但由于这些种间的SSR穿梭标记通常来自于物种基因组的保守区域,检测得到的金花菜多样性并不高,且难以获得大量的有效标记[13 ] .【本研究切入点】由于缺少基因组信息,金花菜SSR标记的开发只能借鉴其近缘物种的基因组进行,制约着金花菜相关工作的有效开展.需找到均匀覆盖金花菜全基因组的分子标记并高通量开发.2021年金花菜的全基因组测序工作顺利完成,针对该物种基因组进行SSR标记的分析与开发成为可能.【拟解决的关键问题】在perl语言环境下,运行微卫星筛选软件MISA(MIcroSAtellite identification tool)的脚本,分别对金花菜、蒺藜苜蓿和紫花苜蓿的基因组FASTA文件进行扫描,对筛选出的简单重复序列进行统计分析. ...
华中和华南的苜蓿与南苜蓿的生产
1
1987
... 【研究意义】金花菜(Medicago polymorpha )属豆科苜蓿属一年生苜蓿[1 ] .金花菜在食用、饲用、药用和绿肥有较高价值[2 ] ,早年金花菜在我国栽培面积达20×104 hm2 (300万亩)[3 ] .近年来,金花菜是我国极具发展前景的多用途豆科牧草[4 ] .金花菜其所在的豆科苜蓿属(Medicago )大约有87个种,包括了豆科模式植物蒺藜苜蓿(Medicago truncatula )和最重要的豆科牧草紫花苜蓿(Medicago sativa )[5 ] .金花菜作为苜蓿属“Polymorpha clade”进化分支的代表性物种,其染色体数目的非整倍体减少(基本染色体数8→7)[6 ] .利用金花菜全基因组测序数据,分析其基因组中简单重复序列的分布特征及与蒺藜苜蓿和紫花苜蓿的异同,对金花菜种质资源遗传多样性和分子标记辅助选育有重要意义.【前人研究进展】简单重复序列(SSR, Simple Sequence Repeats)又称微卫星,为共显性标记,具有扩增稳定、数量丰富、多态性高及特异性强等优势[7 ] 、指纹图谱构建[8 ] 、遗传连锁图谱[9 ] 及QTLs定位等研究[10 ] .利用高通量测序数据开发SSR标记是一种快速、高效、低成本的策略.由于金花菜等一年生苜蓿缺乏基因组序列信息,Eujayl等[11 ] 提出利用豆科模式植物蒺藜苜蓿的ESTs(Expressed sequence tags)序列,开发可用于其它一年生苜蓿的EST-SSR穿梭标记,89%的蒺藜苜蓿EST-SSRs在其他一年生苜蓿上可以跑出条带.Chu等[12 ] 通过对92对蒺藜苜蓿基因组SSR的研究发现,有53%的蒺藜苜蓿基因组SSR标记可以在金花菜上通用.但由于这些种间的SSR穿梭标记通常来自于物种基因组的保守区域,检测得到的金花菜多样性并不高,且难以获得大量的有效标记[13 ] .【本研究切入点】由于缺少基因组信息,金花菜SSR标记的开发只能借鉴其近缘物种的基因组进行,制约着金花菜相关工作的有效开展.需找到均匀覆盖金花菜全基因组的分子标记并高通量开发.2021年金花菜的全基因组测序工作顺利完成,针对该物种基因组进行SSR标记的分析与开发成为可能.【拟解决的关键问题】在perl语言环境下,运行微卫星筛选软件MISA(MIcroSAtellite identification tool)的脚本,分别对金花菜、蒺藜苜蓿和紫花苜蓿的基因组FASTA文件进行扫描,对筛选出的简单重复序列进行统计分析. ...
华中和华南的苜蓿与南苜蓿的生产
1
1987
... 【研究意义】金花菜(Medicago polymorpha )属豆科苜蓿属一年生苜蓿[1 ] .金花菜在食用、饲用、药用和绿肥有较高价值[2 ] ,早年金花菜在我国栽培面积达20×104 hm2 (300万亩)[3 ] .近年来,金花菜是我国极具发展前景的多用途豆科牧草[4 ] .金花菜其所在的豆科苜蓿属(Medicago )大约有87个种,包括了豆科模式植物蒺藜苜蓿(Medicago truncatula )和最重要的豆科牧草紫花苜蓿(Medicago sativa )[5 ] .金花菜作为苜蓿属“Polymorpha clade”进化分支的代表性物种,其染色体数目的非整倍体减少(基本染色体数8→7)[6 ] .利用金花菜全基因组测序数据,分析其基因组中简单重复序列的分布特征及与蒺藜苜蓿和紫花苜蓿的异同,对金花菜种质资源遗传多样性和分子标记辅助选育有重要意义.【前人研究进展】简单重复序列(SSR, Simple Sequence Repeats)又称微卫星,为共显性标记,具有扩增稳定、数量丰富、多态性高及特异性强等优势[7 ] 、指纹图谱构建[8 ] 、遗传连锁图谱[9 ] 及QTLs定位等研究[10 ] .利用高通量测序数据开发SSR标记是一种快速、高效、低成本的策略.由于金花菜等一年生苜蓿缺乏基因组序列信息,Eujayl等[11 ] 提出利用豆科模式植物蒺藜苜蓿的ESTs(Expressed sequence tags)序列,开发可用于其它一年生苜蓿的EST-SSR穿梭标记,89%的蒺藜苜蓿EST-SSRs在其他一年生苜蓿上可以跑出条带.Chu等[12 ] 通过对92对蒺藜苜蓿基因组SSR的研究发现,有53%的蒺藜苜蓿基因组SSR标记可以在金花菜上通用.但由于这些种间的SSR穿梭标记通常来自于物种基因组的保守区域,检测得到的金花菜多样性并不高,且难以获得大量的有效标记[13 ] .【本研究切入点】由于缺少基因组信息,金花菜SSR标记的开发只能借鉴其近缘物种的基因组进行,制约着金花菜相关工作的有效开展.需找到均匀覆盖金花菜全基因组的分子标记并高通量开发.2021年金花菜的全基因组测序工作顺利完成,针对该物种基因组进行SSR标记的分析与开发成为可能.【拟解决的关键问题】在perl语言环境下,运行微卫星筛选软件MISA(MIcroSAtellite identification tool)的脚本,分别对金花菜、蒺藜苜蓿和紫花苜蓿的基因组FASTA文件进行扫描,对筛选出的简单重复序列进行统计分析. ...
扬中金花菜产业发展新模式——南方草业的新亮点
1
2012
... 【研究意义】金花菜(Medicago polymorpha )属豆科苜蓿属一年生苜蓿[1 ] .金花菜在食用、饲用、药用和绿肥有较高价值[2 ] ,早年金花菜在我国栽培面积达20×104 hm2 (300万亩)[3 ] .近年来,金花菜是我国极具发展前景的多用途豆科牧草[4 ] .金花菜其所在的豆科苜蓿属(Medicago )大约有87个种,包括了豆科模式植物蒺藜苜蓿(Medicago truncatula )和最重要的豆科牧草紫花苜蓿(Medicago sativa )[5 ] .金花菜作为苜蓿属“Polymorpha clade”进化分支的代表性物种,其染色体数目的非整倍体减少(基本染色体数8→7)[6 ] .利用金花菜全基因组测序数据,分析其基因组中简单重复序列的分布特征及与蒺藜苜蓿和紫花苜蓿的异同,对金花菜种质资源遗传多样性和分子标记辅助选育有重要意义.【前人研究进展】简单重复序列(SSR, Simple Sequence Repeats)又称微卫星,为共显性标记,具有扩增稳定、数量丰富、多态性高及特异性强等优势[7 ] 、指纹图谱构建[8 ] 、遗传连锁图谱[9 ] 及QTLs定位等研究[10 ] .利用高通量测序数据开发SSR标记是一种快速、高效、低成本的策略.由于金花菜等一年生苜蓿缺乏基因组序列信息,Eujayl等[11 ] 提出利用豆科模式植物蒺藜苜蓿的ESTs(Expressed sequence tags)序列,开发可用于其它一年生苜蓿的EST-SSR穿梭标记,89%的蒺藜苜蓿EST-SSRs在其他一年生苜蓿上可以跑出条带.Chu等[12 ] 通过对92对蒺藜苜蓿基因组SSR的研究发现,有53%的蒺藜苜蓿基因组SSR标记可以在金花菜上通用.但由于这些种间的SSR穿梭标记通常来自于物种基因组的保守区域,检测得到的金花菜多样性并不高,且难以获得大量的有效标记[13 ] .【本研究切入点】由于缺少基因组信息,金花菜SSR标记的开发只能借鉴其近缘物种的基因组进行,制约着金花菜相关工作的有效开展.需找到均匀覆盖金花菜全基因组的分子标记并高通量开发.2021年金花菜的全基因组测序工作顺利完成,针对该物种基因组进行SSR标记的分析与开发成为可能.【拟解决的关键问题】在perl语言环境下,运行微卫星筛选软件MISA(MIcroSAtellite identification tool)的脚本,分别对金花菜、蒺藜苜蓿和紫花苜蓿的基因组FASTA文件进行扫描,对筛选出的简单重复序列进行统计分析. ...
扬中金花菜产业发展新模式——南方草业的新亮点
1
2012
... 【研究意义】金花菜(Medicago polymorpha )属豆科苜蓿属一年生苜蓿[1 ] .金花菜在食用、饲用、药用和绿肥有较高价值[2 ] ,早年金花菜在我国栽培面积达20×104 hm2 (300万亩)[3 ] .近年来,金花菜是我国极具发展前景的多用途豆科牧草[4 ] .金花菜其所在的豆科苜蓿属(Medicago )大约有87个种,包括了豆科模式植物蒺藜苜蓿(Medicago truncatula )和最重要的豆科牧草紫花苜蓿(Medicago sativa )[5 ] .金花菜作为苜蓿属“Polymorpha clade”进化分支的代表性物种,其染色体数目的非整倍体减少(基本染色体数8→7)[6 ] .利用金花菜全基因组测序数据,分析其基因组中简单重复序列的分布特征及与蒺藜苜蓿和紫花苜蓿的异同,对金花菜种质资源遗传多样性和分子标记辅助选育有重要意义.【前人研究进展】简单重复序列(SSR, Simple Sequence Repeats)又称微卫星,为共显性标记,具有扩增稳定、数量丰富、多态性高及特异性强等优势[7 ] 、指纹图谱构建[8 ] 、遗传连锁图谱[9 ] 及QTLs定位等研究[10 ] .利用高通量测序数据开发SSR标记是一种快速、高效、低成本的策略.由于金花菜等一年生苜蓿缺乏基因组序列信息,Eujayl等[11 ] 提出利用豆科模式植物蒺藜苜蓿的ESTs(Expressed sequence tags)序列,开发可用于其它一年生苜蓿的EST-SSR穿梭标记,89%的蒺藜苜蓿EST-SSRs在其他一年生苜蓿上可以跑出条带.Chu等[12 ] 通过对92对蒺藜苜蓿基因组SSR的研究发现,有53%的蒺藜苜蓿基因组SSR标记可以在金花菜上通用.但由于这些种间的SSR穿梭标记通常来自于物种基因组的保守区域,检测得到的金花菜多样性并不高,且难以获得大量的有效标记[13 ] .【本研究切入点】由于缺少基因组信息,金花菜SSR标记的开发只能借鉴其近缘物种的基因组进行,制约着金花菜相关工作的有效开展.需找到均匀覆盖金花菜全基因组的分子标记并高通量开发.2021年金花菜的全基因组测序工作顺利完成,针对该物种基因组进行SSR标记的分析与开发成为可能.【拟解决的关键问题】在perl语言环境下,运行微卫星筛选软件MISA(MIcroSAtellite identification tool)的脚本,分别对金花菜、蒺藜苜蓿和紫花苜蓿的基因组FASTA文件进行扫描,对筛选出的简单重复序列进行统计分析. ...
Phylogeny and character evolution in Medicago (Leguminosae): Evidence from analyses of plastid trnK/matK and nuclear GA3ox1 sequences
1
2010
... 【研究意义】金花菜(Medicago polymorpha )属豆科苜蓿属一年生苜蓿[1 ] .金花菜在食用、饲用、药用和绿肥有较高价值[2 ] ,早年金花菜在我国栽培面积达20×104 hm2 (300万亩)[3 ] .近年来,金花菜是我国极具发展前景的多用途豆科牧草[4 ] .金花菜其所在的豆科苜蓿属(Medicago )大约有87个种,包括了豆科模式植物蒺藜苜蓿(Medicago truncatula )和最重要的豆科牧草紫花苜蓿(Medicago sativa )[5 ] .金花菜作为苜蓿属“Polymorpha clade”进化分支的代表性物种,其染色体数目的非整倍体减少(基本染色体数8→7)[6 ] .利用金花菜全基因组测序数据,分析其基因组中简单重复序列的分布特征及与蒺藜苜蓿和紫花苜蓿的异同,对金花菜种质资源遗传多样性和分子标记辅助选育有重要意义.【前人研究进展】简单重复序列(SSR, Simple Sequence Repeats)又称微卫星,为共显性标记,具有扩增稳定、数量丰富、多态性高及特异性强等优势[7 ] 、指纹图谱构建[8 ] 、遗传连锁图谱[9 ] 及QTLs定位等研究[10 ] .利用高通量测序数据开发SSR标记是一种快速、高效、低成本的策略.由于金花菜等一年生苜蓿缺乏基因组序列信息,Eujayl等[11 ] 提出利用豆科模式植物蒺藜苜蓿的ESTs(Expressed sequence tags)序列,开发可用于其它一年生苜蓿的EST-SSR穿梭标记,89%的蒺藜苜蓿EST-SSRs在其他一年生苜蓿上可以跑出条带.Chu等[12 ] 通过对92对蒺藜苜蓿基因组SSR的研究发现,有53%的蒺藜苜蓿基因组SSR标记可以在金花菜上通用.但由于这些种间的SSR穿梭标记通常来自于物种基因组的保守区域,检测得到的金花菜多样性并不高,且难以获得大量的有效标记[13 ] .【本研究切入点】由于缺少基因组信息,金花菜SSR标记的开发只能借鉴其近缘物种的基因组进行,制约着金花菜相关工作的有效开展.需找到均匀覆盖金花菜全基因组的分子标记并高通量开发.2021年金花菜的全基因组测序工作顺利完成,针对该物种基因组进行SSR标记的分析与开发成为可能.【拟解决的关键问题】在perl语言环境下,运行微卫星筛选软件MISA(MIcroSAtellite identification tool)的脚本,分别对金花菜、蒺藜苜蓿和紫花苜蓿的基因组FASTA文件进行扫描,对筛选出的简单重复序列进行统计分析. ...
Phylogenetic signal variation in the genomes of Medicago (fabaceae)
1
2013
... 【研究意义】金花菜(Medicago polymorpha )属豆科苜蓿属一年生苜蓿[1 ] .金花菜在食用、饲用、药用和绿肥有较高价值[2 ] ,早年金花菜在我国栽培面积达20×104 hm2 (300万亩)[3 ] .近年来,金花菜是我国极具发展前景的多用途豆科牧草[4 ] .金花菜其所在的豆科苜蓿属(Medicago )大约有87个种,包括了豆科模式植物蒺藜苜蓿(Medicago truncatula )和最重要的豆科牧草紫花苜蓿(Medicago sativa )[5 ] .金花菜作为苜蓿属“Polymorpha clade”进化分支的代表性物种,其染色体数目的非整倍体减少(基本染色体数8→7)[6 ] .利用金花菜全基因组测序数据,分析其基因组中简单重复序列的分布特征及与蒺藜苜蓿和紫花苜蓿的异同,对金花菜种质资源遗传多样性和分子标记辅助选育有重要意义.【前人研究进展】简单重复序列(SSR, Simple Sequence Repeats)又称微卫星,为共显性标记,具有扩增稳定、数量丰富、多态性高及特异性强等优势[7 ] 、指纹图谱构建[8 ] 、遗传连锁图谱[9 ] 及QTLs定位等研究[10 ] .利用高通量测序数据开发SSR标记是一种快速、高效、低成本的策略.由于金花菜等一年生苜蓿缺乏基因组序列信息,Eujayl等[11 ] 提出利用豆科模式植物蒺藜苜蓿的ESTs(Expressed sequence tags)序列,开发可用于其它一年生苜蓿的EST-SSR穿梭标记,89%的蒺藜苜蓿EST-SSRs在其他一年生苜蓿上可以跑出条带.Chu等[12 ] 通过对92对蒺藜苜蓿基因组SSR的研究发现,有53%的蒺藜苜蓿基因组SSR标记可以在金花菜上通用.但由于这些种间的SSR穿梭标记通常来自于物种基因组的保守区域,检测得到的金花菜多样性并不高,且难以获得大量的有效标记[13 ] .【本研究切入点】由于缺少基因组信息,金花菜SSR标记的开发只能借鉴其近缘物种的基因组进行,制约着金花菜相关工作的有效开展.需找到均匀覆盖金花菜全基因组的分子标记并高通量开发.2021年金花菜的全基因组测序工作顺利完成,针对该物种基因组进行SSR标记的分析与开发成为可能.【拟解决的关键问题】在perl语言环境下,运行微卫星筛选软件MISA(MIcroSAtellite identification tool)的脚本,分别对金花菜、蒺藜苜蓿和紫花苜蓿的基因组FASTA文件进行扫描,对筛选出的简单重复序列进行统计分析. ...
利用SSR标记对不同耐盐紫花苜蓿遗传多样性分析
1
2019
... 【研究意义】金花菜(Medicago polymorpha )属豆科苜蓿属一年生苜蓿[1 ] .金花菜在食用、饲用、药用和绿肥有较高价值[2 ] ,早年金花菜在我国栽培面积达20×104 hm2 (300万亩)[3 ] .近年来,金花菜是我国极具发展前景的多用途豆科牧草[4 ] .金花菜其所在的豆科苜蓿属(Medicago )大约有87个种,包括了豆科模式植物蒺藜苜蓿(Medicago truncatula )和最重要的豆科牧草紫花苜蓿(Medicago sativa )[5 ] .金花菜作为苜蓿属“Polymorpha clade”进化分支的代表性物种,其染色体数目的非整倍体减少(基本染色体数8→7)[6 ] .利用金花菜全基因组测序数据,分析其基因组中简单重复序列的分布特征及与蒺藜苜蓿和紫花苜蓿的异同,对金花菜种质资源遗传多样性和分子标记辅助选育有重要意义.【前人研究进展】简单重复序列(SSR, Simple Sequence Repeats)又称微卫星,为共显性标记,具有扩增稳定、数量丰富、多态性高及特异性强等优势[7 ] 、指纹图谱构建[8 ] 、遗传连锁图谱[9 ] 及QTLs定位等研究[10 ] .利用高通量测序数据开发SSR标记是一种快速、高效、低成本的策略.由于金花菜等一年生苜蓿缺乏基因组序列信息,Eujayl等[11 ] 提出利用豆科模式植物蒺藜苜蓿的ESTs(Expressed sequence tags)序列,开发可用于其它一年生苜蓿的EST-SSR穿梭标记,89%的蒺藜苜蓿EST-SSRs在其他一年生苜蓿上可以跑出条带.Chu等[12 ] 通过对92对蒺藜苜蓿基因组SSR的研究发现,有53%的蒺藜苜蓿基因组SSR标记可以在金花菜上通用.但由于这些种间的SSR穿梭标记通常来自于物种基因组的保守区域,检测得到的金花菜多样性并不高,且难以获得大量的有效标记[13 ] .【本研究切入点】由于缺少基因组信息,金花菜SSR标记的开发只能借鉴其近缘物种的基因组进行,制约着金花菜相关工作的有效开展.需找到均匀覆盖金花菜全基因组的分子标记并高通量开发.2021年金花菜的全基因组测序工作顺利完成,针对该物种基因组进行SSR标记的分析与开发成为可能.【拟解决的关键问题】在perl语言环境下,运行微卫星筛选软件MISA(MIcroSAtellite identification tool)的脚本,分别对金花菜、蒺藜苜蓿和紫花苜蓿的基因组FASTA文件进行扫描,对筛选出的简单重复序列进行统计分析. ...
利用SSR标记对不同耐盐紫花苜蓿遗传多样性分析
1
2019
... 【研究意义】金花菜(Medicago polymorpha )属豆科苜蓿属一年生苜蓿[1 ] .金花菜在食用、饲用、药用和绿肥有较高价值[2 ] ,早年金花菜在我国栽培面积达20×104 hm2 (300万亩)[3 ] .近年来,金花菜是我国极具发展前景的多用途豆科牧草[4 ] .金花菜其所在的豆科苜蓿属(Medicago )大约有87个种,包括了豆科模式植物蒺藜苜蓿(Medicago truncatula )和最重要的豆科牧草紫花苜蓿(Medicago sativa )[5 ] .金花菜作为苜蓿属“Polymorpha clade”进化分支的代表性物种,其染色体数目的非整倍体减少(基本染色体数8→7)[6 ] .利用金花菜全基因组测序数据,分析其基因组中简单重复序列的分布特征及与蒺藜苜蓿和紫花苜蓿的异同,对金花菜种质资源遗传多样性和分子标记辅助选育有重要意义.【前人研究进展】简单重复序列(SSR, Simple Sequence Repeats)又称微卫星,为共显性标记,具有扩增稳定、数量丰富、多态性高及特异性强等优势[7 ] 、指纹图谱构建[8 ] 、遗传连锁图谱[9 ] 及QTLs定位等研究[10 ] .利用高通量测序数据开发SSR标记是一种快速、高效、低成本的策略.由于金花菜等一年生苜蓿缺乏基因组序列信息,Eujayl等[11 ] 提出利用豆科模式植物蒺藜苜蓿的ESTs(Expressed sequence tags)序列,开发可用于其它一年生苜蓿的EST-SSR穿梭标记,89%的蒺藜苜蓿EST-SSRs在其他一年生苜蓿上可以跑出条带.Chu等[12 ] 通过对92对蒺藜苜蓿基因组SSR的研究发现,有53%的蒺藜苜蓿基因组SSR标记可以在金花菜上通用.但由于这些种间的SSR穿梭标记通常来自于物种基因组的保守区域,检测得到的金花菜多样性并不高,且难以获得大量的有效标记[13 ] .【本研究切入点】由于缺少基因组信息,金花菜SSR标记的开发只能借鉴其近缘物种的基因组进行,制约着金花菜相关工作的有效开展.需找到均匀覆盖金花菜全基因组的分子标记并高通量开发.2021年金花菜的全基因组测序工作顺利完成,针对该物种基因组进行SSR标记的分析与开发成为可能.【拟解决的关键问题】在perl语言环境下,运行微卫星筛选软件MISA(MIcroSAtellite identification tool)的脚本,分别对金花菜、蒺藜苜蓿和紫花苜蓿的基因组FASTA文件进行扫描,对筛选出的简单重复序列进行统计分析. ...
基于荧光毛细管电泳技术的苜蓿SSR指纹图谱构建
1
2020
... 【研究意义】金花菜(Medicago polymorpha )属豆科苜蓿属一年生苜蓿[1 ] .金花菜在食用、饲用、药用和绿肥有较高价值[2 ] ,早年金花菜在我国栽培面积达20×104 hm2 (300万亩)[3 ] .近年来,金花菜是我国极具发展前景的多用途豆科牧草[4 ] .金花菜其所在的豆科苜蓿属(Medicago )大约有87个种,包括了豆科模式植物蒺藜苜蓿(Medicago truncatula )和最重要的豆科牧草紫花苜蓿(Medicago sativa )[5 ] .金花菜作为苜蓿属“Polymorpha clade”进化分支的代表性物种,其染色体数目的非整倍体减少(基本染色体数8→7)[6 ] .利用金花菜全基因组测序数据,分析其基因组中简单重复序列的分布特征及与蒺藜苜蓿和紫花苜蓿的异同,对金花菜种质资源遗传多样性和分子标记辅助选育有重要意义.【前人研究进展】简单重复序列(SSR, Simple Sequence Repeats)又称微卫星,为共显性标记,具有扩增稳定、数量丰富、多态性高及特异性强等优势[7 ] 、指纹图谱构建[8 ] 、遗传连锁图谱[9 ] 及QTLs定位等研究[10 ] .利用高通量测序数据开发SSR标记是一种快速、高效、低成本的策略.由于金花菜等一年生苜蓿缺乏基因组序列信息,Eujayl等[11 ] 提出利用豆科模式植物蒺藜苜蓿的ESTs(Expressed sequence tags)序列,开发可用于其它一年生苜蓿的EST-SSR穿梭标记,89%的蒺藜苜蓿EST-SSRs在其他一年生苜蓿上可以跑出条带.Chu等[12 ] 通过对92对蒺藜苜蓿基因组SSR的研究发现,有53%的蒺藜苜蓿基因组SSR标记可以在金花菜上通用.但由于这些种间的SSR穿梭标记通常来自于物种基因组的保守区域,检测得到的金花菜多样性并不高,且难以获得大量的有效标记[13 ] .【本研究切入点】由于缺少基因组信息,金花菜SSR标记的开发只能借鉴其近缘物种的基因组进行,制约着金花菜相关工作的有效开展.需找到均匀覆盖金花菜全基因组的分子标记并高通量开发.2021年金花菜的全基因组测序工作顺利完成,针对该物种基因组进行SSR标记的分析与开发成为可能.【拟解决的关键问题】在perl语言环境下,运行微卫星筛选软件MISA(MIcroSAtellite identification tool)的脚本,分别对金花菜、蒺藜苜蓿和紫花苜蓿的基因组FASTA文件进行扫描,对筛选出的简单重复序列进行统计分析. ...
基于荧光毛细管电泳技术的苜蓿SSR指纹图谱构建
1
2020
... 【研究意义】金花菜(Medicago polymorpha )属豆科苜蓿属一年生苜蓿[1 ] .金花菜在食用、饲用、药用和绿肥有较高价值[2 ] ,早年金花菜在我国栽培面积达20×104 hm2 (300万亩)[3 ] .近年来,金花菜是我国极具发展前景的多用途豆科牧草[4 ] .金花菜其所在的豆科苜蓿属(Medicago )大约有87个种,包括了豆科模式植物蒺藜苜蓿(Medicago truncatula )和最重要的豆科牧草紫花苜蓿(Medicago sativa )[5 ] .金花菜作为苜蓿属“Polymorpha clade”进化分支的代表性物种,其染色体数目的非整倍体减少(基本染色体数8→7)[6 ] .利用金花菜全基因组测序数据,分析其基因组中简单重复序列的分布特征及与蒺藜苜蓿和紫花苜蓿的异同,对金花菜种质资源遗传多样性和分子标记辅助选育有重要意义.【前人研究进展】简单重复序列(SSR, Simple Sequence Repeats)又称微卫星,为共显性标记,具有扩增稳定、数量丰富、多态性高及特异性强等优势[7 ] 、指纹图谱构建[8 ] 、遗传连锁图谱[9 ] 及QTLs定位等研究[10 ] .利用高通量测序数据开发SSR标记是一种快速、高效、低成本的策略.由于金花菜等一年生苜蓿缺乏基因组序列信息,Eujayl等[11 ] 提出利用豆科模式植物蒺藜苜蓿的ESTs(Expressed sequence tags)序列,开发可用于其它一年生苜蓿的EST-SSR穿梭标记,89%的蒺藜苜蓿EST-SSRs在其他一年生苜蓿上可以跑出条带.Chu等[12 ] 通过对92对蒺藜苜蓿基因组SSR的研究发现,有53%的蒺藜苜蓿基因组SSR标记可以在金花菜上通用.但由于这些种间的SSR穿梭标记通常来自于物种基因组的保守区域,检测得到的金花菜多样性并不高,且难以获得大量的有效标记[13 ] .【本研究切入点】由于缺少基因组信息,金花菜SSR标记的开发只能借鉴其近缘物种的基因组进行,制约着金花菜相关工作的有效开展.需找到均匀覆盖金花菜全基因组的分子标记并高通量开发.2021年金花菜的全基因组测序工作顺利完成,针对该物种基因组进行SSR标记的分析与开发成为可能.【拟解决的关键问题】在perl语言环境下,运行微卫星筛选软件MISA(MIcroSAtellite identification tool)的脚本,分别对金花菜、蒺藜苜蓿和紫花苜蓿的基因组FASTA文件进行扫描,对筛选出的简单重复序列进行统计分析. ...
四倍体紫花苜蓿遗传图谱的初步构建
1
2015
... 【研究意义】金花菜(Medicago polymorpha )属豆科苜蓿属一年生苜蓿[1 ] .金花菜在食用、饲用、药用和绿肥有较高价值[2 ] ,早年金花菜在我国栽培面积达20×104 hm2 (300万亩)[3 ] .近年来,金花菜是我国极具发展前景的多用途豆科牧草[4 ] .金花菜其所在的豆科苜蓿属(Medicago )大约有87个种,包括了豆科模式植物蒺藜苜蓿(Medicago truncatula )和最重要的豆科牧草紫花苜蓿(Medicago sativa )[5 ] .金花菜作为苜蓿属“Polymorpha clade”进化分支的代表性物种,其染色体数目的非整倍体减少(基本染色体数8→7)[6 ] .利用金花菜全基因组测序数据,分析其基因组中简单重复序列的分布特征及与蒺藜苜蓿和紫花苜蓿的异同,对金花菜种质资源遗传多样性和分子标记辅助选育有重要意义.【前人研究进展】简单重复序列(SSR, Simple Sequence Repeats)又称微卫星,为共显性标记,具有扩增稳定、数量丰富、多态性高及特异性强等优势[7 ] 、指纹图谱构建[8 ] 、遗传连锁图谱[9 ] 及QTLs定位等研究[10 ] .利用高通量测序数据开发SSR标记是一种快速、高效、低成本的策略.由于金花菜等一年生苜蓿缺乏基因组序列信息,Eujayl等[11 ] 提出利用豆科模式植物蒺藜苜蓿的ESTs(Expressed sequence tags)序列,开发可用于其它一年生苜蓿的EST-SSR穿梭标记,89%的蒺藜苜蓿EST-SSRs在其他一年生苜蓿上可以跑出条带.Chu等[12 ] 通过对92对蒺藜苜蓿基因组SSR的研究发现,有53%的蒺藜苜蓿基因组SSR标记可以在金花菜上通用.但由于这些种间的SSR穿梭标记通常来自于物种基因组的保守区域,检测得到的金花菜多样性并不高,且难以获得大量的有效标记[13 ] .【本研究切入点】由于缺少基因组信息,金花菜SSR标记的开发只能借鉴其近缘物种的基因组进行,制约着金花菜相关工作的有效开展.需找到均匀覆盖金花菜全基因组的分子标记并高通量开发.2021年金花菜的全基因组测序工作顺利完成,针对该物种基因组进行SSR标记的分析与开发成为可能.【拟解决的关键问题】在perl语言环境下,运行微卫星筛选软件MISA(MIcroSAtellite identification tool)的脚本,分别对金花菜、蒺藜苜蓿和紫花苜蓿的基因组FASTA文件进行扫描,对筛选出的简单重复序列进行统计分析. ...
四倍体紫花苜蓿遗传图谱的初步构建
1
2015
... 【研究意义】金花菜(Medicago polymorpha )属豆科苜蓿属一年生苜蓿[1 ] .金花菜在食用、饲用、药用和绿肥有较高价值[2 ] ,早年金花菜在我国栽培面积达20×104 hm2 (300万亩)[3 ] .近年来,金花菜是我国极具发展前景的多用途豆科牧草[4 ] .金花菜其所在的豆科苜蓿属(Medicago )大约有87个种,包括了豆科模式植物蒺藜苜蓿(Medicago truncatula )和最重要的豆科牧草紫花苜蓿(Medicago sativa )[5 ] .金花菜作为苜蓿属“Polymorpha clade”进化分支的代表性物种,其染色体数目的非整倍体减少(基本染色体数8→7)[6 ] .利用金花菜全基因组测序数据,分析其基因组中简单重复序列的分布特征及与蒺藜苜蓿和紫花苜蓿的异同,对金花菜种质资源遗传多样性和分子标记辅助选育有重要意义.【前人研究进展】简单重复序列(SSR, Simple Sequence Repeats)又称微卫星,为共显性标记,具有扩增稳定、数量丰富、多态性高及特异性强等优势[7 ] 、指纹图谱构建[8 ] 、遗传连锁图谱[9 ] 及QTLs定位等研究[10 ] .利用高通量测序数据开发SSR标记是一种快速、高效、低成本的策略.由于金花菜等一年生苜蓿缺乏基因组序列信息,Eujayl等[11 ] 提出利用豆科模式植物蒺藜苜蓿的ESTs(Expressed sequence tags)序列,开发可用于其它一年生苜蓿的EST-SSR穿梭标记,89%的蒺藜苜蓿EST-SSRs在其他一年生苜蓿上可以跑出条带.Chu等[12 ] 通过对92对蒺藜苜蓿基因组SSR的研究发现,有53%的蒺藜苜蓿基因组SSR标记可以在金花菜上通用.但由于这些种间的SSR穿梭标记通常来自于物种基因组的保守区域,检测得到的金花菜多样性并不高,且难以获得大量的有效标记[13 ] .【本研究切入点】由于缺少基因组信息,金花菜SSR标记的开发只能借鉴其近缘物种的基因组进行,制约着金花菜相关工作的有效开展.需找到均匀覆盖金花菜全基因组的分子标记并高通量开发.2021年金花菜的全基因组测序工作顺利完成,针对该物种基因组进行SSR标记的分析与开发成为可能.【拟解决的关键问题】在perl语言环境下,运行微卫星筛选软件MISA(MIcroSAtellite identification tool)的脚本,分别对金花菜、蒺藜苜蓿和紫花苜蓿的基因组FASTA文件进行扫描,对筛选出的简单重复序列进行统计分析. ...
紫花苜蓿细胞质雄性不育恢复基因的初步定位
1
2018
... 【研究意义】金花菜(Medicago polymorpha )属豆科苜蓿属一年生苜蓿[1 ] .金花菜在食用、饲用、药用和绿肥有较高价值[2 ] ,早年金花菜在我国栽培面积达20×104 hm2 (300万亩)[3 ] .近年来,金花菜是我国极具发展前景的多用途豆科牧草[4 ] .金花菜其所在的豆科苜蓿属(Medicago )大约有87个种,包括了豆科模式植物蒺藜苜蓿(Medicago truncatula )和最重要的豆科牧草紫花苜蓿(Medicago sativa )[5 ] .金花菜作为苜蓿属“Polymorpha clade”进化分支的代表性物种,其染色体数目的非整倍体减少(基本染色体数8→7)[6 ] .利用金花菜全基因组测序数据,分析其基因组中简单重复序列的分布特征及与蒺藜苜蓿和紫花苜蓿的异同,对金花菜种质资源遗传多样性和分子标记辅助选育有重要意义.【前人研究进展】简单重复序列(SSR, Simple Sequence Repeats)又称微卫星,为共显性标记,具有扩增稳定、数量丰富、多态性高及特异性强等优势[7 ] 、指纹图谱构建[8 ] 、遗传连锁图谱[9 ] 及QTLs定位等研究[10 ] .利用高通量测序数据开发SSR标记是一种快速、高效、低成本的策略.由于金花菜等一年生苜蓿缺乏基因组序列信息,Eujayl等[11 ] 提出利用豆科模式植物蒺藜苜蓿的ESTs(Expressed sequence tags)序列,开发可用于其它一年生苜蓿的EST-SSR穿梭标记,89%的蒺藜苜蓿EST-SSRs在其他一年生苜蓿上可以跑出条带.Chu等[12 ] 通过对92对蒺藜苜蓿基因组SSR的研究发现,有53%的蒺藜苜蓿基因组SSR标记可以在金花菜上通用.但由于这些种间的SSR穿梭标记通常来自于物种基因组的保守区域,检测得到的金花菜多样性并不高,且难以获得大量的有效标记[13 ] .【本研究切入点】由于缺少基因组信息,金花菜SSR标记的开发只能借鉴其近缘物种的基因组进行,制约着金花菜相关工作的有效开展.需找到均匀覆盖金花菜全基因组的分子标记并高通量开发.2021年金花菜的全基因组测序工作顺利完成,针对该物种基因组进行SSR标记的分析与开发成为可能.【拟解决的关键问题】在perl语言环境下,运行微卫星筛选软件MISA(MIcroSAtellite identification tool)的脚本,分别对金花菜、蒺藜苜蓿和紫花苜蓿的基因组FASTA文件进行扫描,对筛选出的简单重复序列进行统计分析. ...
紫花苜蓿细胞质雄性不育恢复基因的初步定位
1
2018
... 【研究意义】金花菜(Medicago polymorpha )属豆科苜蓿属一年生苜蓿[1 ] .金花菜在食用、饲用、药用和绿肥有较高价值[2 ] ,早年金花菜在我国栽培面积达20×104 hm2 (300万亩)[3 ] .近年来,金花菜是我国极具发展前景的多用途豆科牧草[4 ] .金花菜其所在的豆科苜蓿属(Medicago )大约有87个种,包括了豆科模式植物蒺藜苜蓿(Medicago truncatula )和最重要的豆科牧草紫花苜蓿(Medicago sativa )[5 ] .金花菜作为苜蓿属“Polymorpha clade”进化分支的代表性物种,其染色体数目的非整倍体减少(基本染色体数8→7)[6 ] .利用金花菜全基因组测序数据,分析其基因组中简单重复序列的分布特征及与蒺藜苜蓿和紫花苜蓿的异同,对金花菜种质资源遗传多样性和分子标记辅助选育有重要意义.【前人研究进展】简单重复序列(SSR, Simple Sequence Repeats)又称微卫星,为共显性标记,具有扩增稳定、数量丰富、多态性高及特异性强等优势[7 ] 、指纹图谱构建[8 ] 、遗传连锁图谱[9 ] 及QTLs定位等研究[10 ] .利用高通量测序数据开发SSR标记是一种快速、高效、低成本的策略.由于金花菜等一年生苜蓿缺乏基因组序列信息,Eujayl等[11 ] 提出利用豆科模式植物蒺藜苜蓿的ESTs(Expressed sequence tags)序列,开发可用于其它一年生苜蓿的EST-SSR穿梭标记,89%的蒺藜苜蓿EST-SSRs在其他一年生苜蓿上可以跑出条带.Chu等[12 ] 通过对92对蒺藜苜蓿基因组SSR的研究发现,有53%的蒺藜苜蓿基因组SSR标记可以在金花菜上通用.但由于这些种间的SSR穿梭标记通常来自于物种基因组的保守区域,检测得到的金花菜多样性并不高,且难以获得大量的有效标记[13 ] .【本研究切入点】由于缺少基因组信息,金花菜SSR标记的开发只能借鉴其近缘物种的基因组进行,制约着金花菜相关工作的有效开展.需找到均匀覆盖金花菜全基因组的分子标记并高通量开发.2021年金花菜的全基因组测序工作顺利完成,针对该物种基因组进行SSR标记的分析与开发成为可能.【拟解决的关键问题】在perl语言环境下,运行微卫星筛选软件MISA(MIcroSAtellite identification tool)的脚本,分别对金花菜、蒺藜苜蓿和紫花苜蓿的基因组FASTA文件进行扫描,对筛选出的简单重复序列进行统计分析. ...
Medicago truncatula EST-SSRs reveal cross-species genetic markers for Medicago spp
1
2004
... 【研究意义】金花菜(Medicago polymorpha )属豆科苜蓿属一年生苜蓿[1 ] .金花菜在食用、饲用、药用和绿肥有较高价值[2 ] ,早年金花菜在我国栽培面积达20×104 hm2 (300万亩)[3 ] .近年来,金花菜是我国极具发展前景的多用途豆科牧草[4 ] .金花菜其所在的豆科苜蓿属(Medicago )大约有87个种,包括了豆科模式植物蒺藜苜蓿(Medicago truncatula )和最重要的豆科牧草紫花苜蓿(Medicago sativa )[5 ] .金花菜作为苜蓿属“Polymorpha clade”进化分支的代表性物种,其染色体数目的非整倍体减少(基本染色体数8→7)[6 ] .利用金花菜全基因组测序数据,分析其基因组中简单重复序列的分布特征及与蒺藜苜蓿和紫花苜蓿的异同,对金花菜种质资源遗传多样性和分子标记辅助选育有重要意义.【前人研究进展】简单重复序列(SSR, Simple Sequence Repeats)又称微卫星,为共显性标记,具有扩增稳定、数量丰富、多态性高及特异性强等优势[7 ] 、指纹图谱构建[8 ] 、遗传连锁图谱[9 ] 及QTLs定位等研究[10 ] .利用高通量测序数据开发SSR标记是一种快速、高效、低成本的策略.由于金花菜等一年生苜蓿缺乏基因组序列信息,Eujayl等[11 ] 提出利用豆科模式植物蒺藜苜蓿的ESTs(Expressed sequence tags)序列,开发可用于其它一年生苜蓿的EST-SSR穿梭标记,89%的蒺藜苜蓿EST-SSRs在其他一年生苜蓿上可以跑出条带.Chu等[12 ] 通过对92对蒺藜苜蓿基因组SSR的研究发现,有53%的蒺藜苜蓿基因组SSR标记可以在金花菜上通用.但由于这些种间的SSR穿梭标记通常来自于物种基因组的保守区域,检测得到的金花菜多样性并不高,且难以获得大量的有效标记[13 ] .【本研究切入点】由于缺少基因组信息,金花菜SSR标记的开发只能借鉴其近缘物种的基因组进行,制约着金花菜相关工作的有效开展.需找到均匀覆盖金花菜全基因组的分子标记并高通量开发.2021年金花菜的全基因组测序工作顺利完成,针对该物种基因组进行SSR标记的分析与开发成为可能.【拟解决的关键问题】在perl语言环境下,运行微卫星筛选软件MISA(MIcroSAtellite identification tool)的脚本,分别对金花菜、蒺藜苜蓿和紫花苜蓿的基因组FASTA文件进行扫描,对筛选出的简单重复序列进行统计分析. ...
Cross-species amplification of 92 microsatellites of Medicago truncatula
1
2010
... 【研究意义】金花菜(Medicago polymorpha )属豆科苜蓿属一年生苜蓿[1 ] .金花菜在食用、饲用、药用和绿肥有较高价值[2 ] ,早年金花菜在我国栽培面积达20×104 hm2 (300万亩)[3 ] .近年来,金花菜是我国极具发展前景的多用途豆科牧草[4 ] .金花菜其所在的豆科苜蓿属(Medicago )大约有87个种,包括了豆科模式植物蒺藜苜蓿(Medicago truncatula )和最重要的豆科牧草紫花苜蓿(Medicago sativa )[5 ] .金花菜作为苜蓿属“Polymorpha clade”进化分支的代表性物种,其染色体数目的非整倍体减少(基本染色体数8→7)[6 ] .利用金花菜全基因组测序数据,分析其基因组中简单重复序列的分布特征及与蒺藜苜蓿和紫花苜蓿的异同,对金花菜种质资源遗传多样性和分子标记辅助选育有重要意义.【前人研究进展】简单重复序列(SSR, Simple Sequence Repeats)又称微卫星,为共显性标记,具有扩增稳定、数量丰富、多态性高及特异性强等优势[7 ] 、指纹图谱构建[8 ] 、遗传连锁图谱[9 ] 及QTLs定位等研究[10 ] .利用高通量测序数据开发SSR标记是一种快速、高效、低成本的策略.由于金花菜等一年生苜蓿缺乏基因组序列信息,Eujayl等[11 ] 提出利用豆科模式植物蒺藜苜蓿的ESTs(Expressed sequence tags)序列,开发可用于其它一年生苜蓿的EST-SSR穿梭标记,89%的蒺藜苜蓿EST-SSRs在其他一年生苜蓿上可以跑出条带.Chu等[12 ] 通过对92对蒺藜苜蓿基因组SSR的研究发现,有53%的蒺藜苜蓿基因组SSR标记可以在金花菜上通用.但由于这些种间的SSR穿梭标记通常来自于物种基因组的保守区域,检测得到的金花菜多样性并不高,且难以获得大量的有效标记[13 ] .【本研究切入点】由于缺少基因组信息,金花菜SSR标记的开发只能借鉴其近缘物种的基因组进行,制约着金花菜相关工作的有效开展.需找到均匀覆盖金花菜全基因组的分子标记并高通量开发.2021年金花菜的全基因组测序工作顺利完成,针对该物种基因组进行SSR标记的分析与开发成为可能.【拟解决的关键问题】在perl语言环境下,运行微卫星筛选软件MISA(MIcroSAtellite identification tool)的脚本,分别对金花菜、蒺藜苜蓿和紫花苜蓿的基因组FASTA文件进行扫描,对筛选出的简单重复序列进行统计分析. ...
聚丙烯酰胺凝胶电泳检测南苜蓿SSR标记
1
2015
... 【研究意义】金花菜(Medicago polymorpha )属豆科苜蓿属一年生苜蓿[1 ] .金花菜在食用、饲用、药用和绿肥有较高价值[2 ] ,早年金花菜在我国栽培面积达20×104 hm2 (300万亩)[3 ] .近年来,金花菜是我国极具发展前景的多用途豆科牧草[4 ] .金花菜其所在的豆科苜蓿属(Medicago )大约有87个种,包括了豆科模式植物蒺藜苜蓿(Medicago truncatula )和最重要的豆科牧草紫花苜蓿(Medicago sativa )[5 ] .金花菜作为苜蓿属“Polymorpha clade”进化分支的代表性物种,其染色体数目的非整倍体减少(基本染色体数8→7)[6 ] .利用金花菜全基因组测序数据,分析其基因组中简单重复序列的分布特征及与蒺藜苜蓿和紫花苜蓿的异同,对金花菜种质资源遗传多样性和分子标记辅助选育有重要意义.【前人研究进展】简单重复序列(SSR, Simple Sequence Repeats)又称微卫星,为共显性标记,具有扩增稳定、数量丰富、多态性高及特异性强等优势[7 ] 、指纹图谱构建[8 ] 、遗传连锁图谱[9 ] 及QTLs定位等研究[10 ] .利用高通量测序数据开发SSR标记是一种快速、高效、低成本的策略.由于金花菜等一年生苜蓿缺乏基因组序列信息,Eujayl等[11 ] 提出利用豆科模式植物蒺藜苜蓿的ESTs(Expressed sequence tags)序列,开发可用于其它一年生苜蓿的EST-SSR穿梭标记,89%的蒺藜苜蓿EST-SSRs在其他一年生苜蓿上可以跑出条带.Chu等[12 ] 通过对92对蒺藜苜蓿基因组SSR的研究发现,有53%的蒺藜苜蓿基因组SSR标记可以在金花菜上通用.但由于这些种间的SSR穿梭标记通常来自于物种基因组的保守区域,检测得到的金花菜多样性并不高,且难以获得大量的有效标记[13 ] .【本研究切入点】由于缺少基因组信息,金花菜SSR标记的开发只能借鉴其近缘物种的基因组进行,制约着金花菜相关工作的有效开展.需找到均匀覆盖金花菜全基因组的分子标记并高通量开发.2021年金花菜的全基因组测序工作顺利完成,针对该物种基因组进行SSR标记的分析与开发成为可能.【拟解决的关键问题】在perl语言环境下,运行微卫星筛选软件MISA(MIcroSAtellite identification tool)的脚本,分别对金花菜、蒺藜苜蓿和紫花苜蓿的基因组FASTA文件进行扫描,对筛选出的简单重复序列进行统计分析. ...
聚丙烯酰胺凝胶电泳检测南苜蓿SSR标记
1
2015
... 【研究意义】金花菜(Medicago polymorpha )属豆科苜蓿属一年生苜蓿[1 ] .金花菜在食用、饲用、药用和绿肥有较高价值[2 ] ,早年金花菜在我国栽培面积达20×104 hm2 (300万亩)[3 ] .近年来,金花菜是我国极具发展前景的多用途豆科牧草[4 ] .金花菜其所在的豆科苜蓿属(Medicago )大约有87个种,包括了豆科模式植物蒺藜苜蓿(Medicago truncatula )和最重要的豆科牧草紫花苜蓿(Medicago sativa )[5 ] .金花菜作为苜蓿属“Polymorpha clade”进化分支的代表性物种,其染色体数目的非整倍体减少(基本染色体数8→7)[6 ] .利用金花菜全基因组测序数据,分析其基因组中简单重复序列的分布特征及与蒺藜苜蓿和紫花苜蓿的异同,对金花菜种质资源遗传多样性和分子标记辅助选育有重要意义.【前人研究进展】简单重复序列(SSR, Simple Sequence Repeats)又称微卫星,为共显性标记,具有扩增稳定、数量丰富、多态性高及特异性强等优势[7 ] 、指纹图谱构建[8 ] 、遗传连锁图谱[9 ] 及QTLs定位等研究[10 ] .利用高通量测序数据开发SSR标记是一种快速、高效、低成本的策略.由于金花菜等一年生苜蓿缺乏基因组序列信息,Eujayl等[11 ] 提出利用豆科模式植物蒺藜苜蓿的ESTs(Expressed sequence tags)序列,开发可用于其它一年生苜蓿的EST-SSR穿梭标记,89%的蒺藜苜蓿EST-SSRs在其他一年生苜蓿上可以跑出条带.Chu等[12 ] 通过对92对蒺藜苜蓿基因组SSR的研究发现,有53%的蒺藜苜蓿基因组SSR标记可以在金花菜上通用.但由于这些种间的SSR穿梭标记通常来自于物种基因组的保守区域,检测得到的金花菜多样性并不高,且难以获得大量的有效标记[13 ] .【本研究切入点】由于缺少基因组信息,金花菜SSR标记的开发只能借鉴其近缘物种的基因组进行,制约着金花菜相关工作的有效开展.需找到均匀覆盖金花菜全基因组的分子标记并高通量开发.2021年金花菜的全基因组测序工作顺利完成,针对该物种基因组进行SSR标记的分析与开发成为可能.【拟解决的关键问题】在perl语言环境下,运行微卫星筛选软件MISA(MIcroSAtellite identification tool)的脚本,分别对金花菜、蒺藜苜蓿和紫花苜蓿的基因组FASTA文件进行扫描,对筛选出的简单重复序列进行统计分析. ...
The genome of Medicago polymorpha provides insights into its edibility and nutritional value as a vegetable and forage legume
2
2021
... 金花菜基因组从国家基因组科学数据中心(National Genomics Data Center)数据库下载(https://bigd.big.ac.cn/gsa/s/q0VtV4XI )[14 ] ;蒺藜苜蓿基因组(Mt 5.0)从美国国立生物技术信息中心(National Center for Biotechnology Information)数据库下载(https://www.ncbi.nlm.nih.gov/genome/6?genome_assembly_id=406060 )[15 ] ;紫花苜蓿基因组从Figshare科学数据共享平台下载(https://figshare.com/articles/dataset/Medicago_sativa_genome_and_annotation_files/12623960 )[16 ] ,3种苜蓿基因组所有序列均以FASTA文件格式保存.表1 ...
... SSR序列长度<12 bp时SSR标记的多态性表现极低;序列长度在12~20 bp之间时标记多态性适中;≥20 bp时具有较高多态性,是理想的标记位点[18 ] .基因组中存在着大量的重复序列,从进化角度看,物种间重复序列的差异是自然选择的结果,因此鉴定SSR在基因组中的分布特征有重要意义[19 ] .金花菜、蒺藜苜蓿和紫花苜蓿是苜蓿属的不同种,其中金花菜和蒺藜苜蓿属于一年生苜蓿,紫花苜蓿属于多年生苜蓿,3种苜蓿基因组有很强的的共线性关系[14 ] .研究发现,金花菜基因组SSR的分布密度为428个/Mb,明显低于蒺藜苜蓿的分布密度(564个/Mb)以及紫花苜蓿的分布密度(478个/Mb).Varshney等[20 ] 研究认为,SSR分布密度之所以出现差异,除了物种间差异因素外,还与测序数据深度、序列拼接数据质量及SSR位点查找软件以及SSR搜索标准不同有关.研究选用主流的微卫星筛选软件MISA(MIcroSAtellite identification tool),在相同设置条件下分析了这3种苜蓿间差异.金花菜、蒺藜苜蓿和紫花苜蓿的测序深度分别为117X、109X和153X,均为二代+三代测序组装的高质量基因组,结果比较能真实发映出物种间的差异.金花菜基因组SSR的分布密度较低,可能与金花菜染色体数目少有关. ...
Whole-genome landscape of Medicago truncatula symbiotic genes
1
2018
... 金花菜基因组从国家基因组科学数据中心(National Genomics Data Center)数据库下载(https://bigd.big.ac.cn/gsa/s/q0VtV4XI )[14 ] ;蒺藜苜蓿基因组(Mt 5.0)从美国国立生物技术信息中心(National Center for Biotechnology Information)数据库下载(https://www.ncbi.nlm.nih.gov/genome/6?genome_assembly_id=406060 )[15 ] ;紫花苜蓿基因组从Figshare科学数据共享平台下载(https://figshare.com/articles/dataset/Medicago_sativa_genome_and_annotation_files/12623960 )[16 ] ,3种苜蓿基因组所有序列均以FASTA文件格式保存.表1 ...
The Chromosome-Level Genome Sequence of the Autotetraploid Alfalfa and Resequencing of Core Germplasms Provide Genomic Resources for Alfalfa Research
1
2020
... 金花菜基因组从国家基因组科学数据中心(National Genomics Data Center)数据库下载(https://bigd.big.ac.cn/gsa/s/q0VtV4XI )[14 ] ;蒺藜苜蓿基因组(Mt 5.0)从美国国立生物技术信息中心(National Center for Biotechnology Information)数据库下载(https://www.ncbi.nlm.nih.gov/genome/6?genome_assembly_id=406060 )[15 ] ;紫花苜蓿基因组从Figshare科学数据共享平台下载(https://figshare.com/articles/dataset/Medicago_sativa_genome_and_annotation_files/12623960 )[16 ] ,3种苜蓿基因组所有序列均以FASTA文件格式保存.表1 ...
MISA-web: a web server for microsatellite prediction
1
2017
... 使用微卫星检索工具MISA[17 ] (https://webblast.ipk-gatersleben.de/misa/ )执行命令perl misa.pl genome.fasta,对3种苜蓿全基因组进行扫描,筛选符合条件的简单重复序列.筛选标准为MISA软件的默认值:单核苷酸重复次数在10次及以上,二核苷酸重复次数在6次及以上,三至六核苷酸重复次数在5次及以上,复合型SSR的检索条件是2个SSR片段间的距离低于100 bp.将生成的数据采用Excel软件整理,对序列特征进行分析并绘制图表. ...
大花序桉基因组SSR的分布特征及序列分析
2
2021
... SSR序列长度<12 bp时SSR标记的多态性表现极低;序列长度在12~20 bp之间时标记多态性适中;≥20 bp时具有较高多态性,是理想的标记位点[18 ] .基因组中存在着大量的重复序列,从进化角度看,物种间重复序列的差异是自然选择的结果,因此鉴定SSR在基因组中的分布特征有重要意义[19 ] .金花菜、蒺藜苜蓿和紫花苜蓿是苜蓿属的不同种,其中金花菜和蒺藜苜蓿属于一年生苜蓿,紫花苜蓿属于多年生苜蓿,3种苜蓿基因组有很强的的共线性关系[14 ] .研究发现,金花菜基因组SSR的分布密度为428个/Mb,明显低于蒺藜苜蓿的分布密度(564个/Mb)以及紫花苜蓿的分布密度(478个/Mb).Varshney等[20 ] 研究认为,SSR分布密度之所以出现差异,除了物种间差异因素外,还与测序数据深度、序列拼接数据质量及SSR位点查找软件以及SSR搜索标准不同有关.研究选用主流的微卫星筛选软件MISA(MIcroSAtellite identification tool),在相同设置条件下分析了这3种苜蓿间差异.金花菜、蒺藜苜蓿和紫花苜蓿的测序深度分别为117X、109X和153X,均为二代+三代测序组装的高质量基因组,结果比较能真实发映出物种间的差异.金花菜基因组SSR的分布密度较低,可能与金花菜染色体数目少有关. ...
... 单核苷酸、二核苷酸和三核苷酸重复单元是绝大多植物基因组SSR序列中优势重复单元[18 ,21 ] .研究发现,金花菜基因组SSR中,单、二和三核苷酸重复单元类型分别占基因组SSR数量的75.58%、15.31%和7.94%,其后依次是,四、五和六核苷酸重复单元,与蒺藜苜蓿观测到的结果相一致.与金花菜和蒺藜苜蓿相比,紫花苜蓿的六核苷酸重复单元数量多于五核苷酸重复单元,且单核苷酸重复单元类型的SSR数量相对较少,这可能与紫花苜蓿是同源四倍体,异花授粉导致遗传变异更为丰富有关. ...
大花序桉基因组SSR的分布特征及序列分析
2
2021
... SSR序列长度<12 bp时SSR标记的多态性表现极低;序列长度在12~20 bp之间时标记多态性适中;≥20 bp时具有较高多态性,是理想的标记位点[18 ] .基因组中存在着大量的重复序列,从进化角度看,物种间重复序列的差异是自然选择的结果,因此鉴定SSR在基因组中的分布特征有重要意义[19 ] .金花菜、蒺藜苜蓿和紫花苜蓿是苜蓿属的不同种,其中金花菜和蒺藜苜蓿属于一年生苜蓿,紫花苜蓿属于多年生苜蓿,3种苜蓿基因组有很强的的共线性关系[14 ] .研究发现,金花菜基因组SSR的分布密度为428个/Mb,明显低于蒺藜苜蓿的分布密度(564个/Mb)以及紫花苜蓿的分布密度(478个/Mb).Varshney等[20 ] 研究认为,SSR分布密度之所以出现差异,除了物种间差异因素外,还与测序数据深度、序列拼接数据质量及SSR位点查找软件以及SSR搜索标准不同有关.研究选用主流的微卫星筛选软件MISA(MIcroSAtellite identification tool),在相同设置条件下分析了这3种苜蓿间差异.金花菜、蒺藜苜蓿和紫花苜蓿的测序深度分别为117X、109X和153X,均为二代+三代测序组装的高质量基因组,结果比较能真实发映出物种间的差异.金花菜基因组SSR的分布密度较低,可能与金花菜染色体数目少有关. ...
... 单核苷酸、二核苷酸和三核苷酸重复单元是绝大多植物基因组SSR序列中优势重复单元[18 ,21 ] .研究发现,金花菜基因组SSR中,单、二和三核苷酸重复单元类型分别占基因组SSR数量的75.58%、15.31%和7.94%,其后依次是,四、五和六核苷酸重复单元,与蒺藜苜蓿观测到的结果相一致.与金花菜和蒺藜苜蓿相比,紫花苜蓿的六核苷酸重复单元数量多于五核苷酸重复单元,且单核苷酸重复单元类型的SSR数量相对较少,这可能与紫花苜蓿是同源四倍体,异花授粉导致遗传变异更为丰富有关. ...
重复序列对植物基因组大小进化的影响
1
2021
... SSR序列长度<12 bp时SSR标记的多态性表现极低;序列长度在12~20 bp之间时标记多态性适中;≥20 bp时具有较高多态性,是理想的标记位点[18 ] .基因组中存在着大量的重复序列,从进化角度看,物种间重复序列的差异是自然选择的结果,因此鉴定SSR在基因组中的分布特征有重要意义[19 ] .金花菜、蒺藜苜蓿和紫花苜蓿是苜蓿属的不同种,其中金花菜和蒺藜苜蓿属于一年生苜蓿,紫花苜蓿属于多年生苜蓿,3种苜蓿基因组有很强的的共线性关系[14 ] .研究发现,金花菜基因组SSR的分布密度为428个/Mb,明显低于蒺藜苜蓿的分布密度(564个/Mb)以及紫花苜蓿的分布密度(478个/Mb).Varshney等[20 ] 研究认为,SSR分布密度之所以出现差异,除了物种间差异因素外,还与测序数据深度、序列拼接数据质量及SSR位点查找软件以及SSR搜索标准不同有关.研究选用主流的微卫星筛选软件MISA(MIcroSAtellite identification tool),在相同设置条件下分析了这3种苜蓿间差异.金花菜、蒺藜苜蓿和紫花苜蓿的测序深度分别为117X、109X和153X,均为二代+三代测序组装的高质量基因组,结果比较能真实发映出物种间的差异.金花菜基因组SSR的分布密度较低,可能与金花菜染色体数目少有关. ...
重复序列对植物基因组大小进化的影响
1
2021
... SSR序列长度<12 bp时SSR标记的多态性表现极低;序列长度在12~20 bp之间时标记多态性适中;≥20 bp时具有较高多态性,是理想的标记位点[18 ] .基因组中存在着大量的重复序列,从进化角度看,物种间重复序列的差异是自然选择的结果,因此鉴定SSR在基因组中的分布特征有重要意义[19 ] .金花菜、蒺藜苜蓿和紫花苜蓿是苜蓿属的不同种,其中金花菜和蒺藜苜蓿属于一年生苜蓿,紫花苜蓿属于多年生苜蓿,3种苜蓿基因组有很强的的共线性关系[14 ] .研究发现,金花菜基因组SSR的分布密度为428个/Mb,明显低于蒺藜苜蓿的分布密度(564个/Mb)以及紫花苜蓿的分布密度(478个/Mb).Varshney等[20 ] 研究认为,SSR分布密度之所以出现差异,除了物种间差异因素外,还与测序数据深度、序列拼接数据质量及SSR位点查找软件以及SSR搜索标准不同有关.研究选用主流的微卫星筛选软件MISA(MIcroSAtellite identification tool),在相同设置条件下分析了这3种苜蓿间差异.金花菜、蒺藜苜蓿和紫花苜蓿的测序深度分别为117X、109X和153X,均为二代+三代测序组装的高质量基因组,结果比较能真实发映出物种间的差异.金花菜基因组SSR的分布密度较低,可能与金花菜染色体数目少有关. ...
Genic microsatellite markers in plants: Features and applications
1
2005
... SSR序列长度<12 bp时SSR标记的多态性表现极低;序列长度在12~20 bp之间时标记多态性适中;≥20 bp时具有较高多态性,是理想的标记位点[18 ] .基因组中存在着大量的重复序列,从进化角度看,物种间重复序列的差异是自然选择的结果,因此鉴定SSR在基因组中的分布特征有重要意义[19 ] .金花菜、蒺藜苜蓿和紫花苜蓿是苜蓿属的不同种,其中金花菜和蒺藜苜蓿属于一年生苜蓿,紫花苜蓿属于多年生苜蓿,3种苜蓿基因组有很强的的共线性关系[14 ] .研究发现,金花菜基因组SSR的分布密度为428个/Mb,明显低于蒺藜苜蓿的分布密度(564个/Mb)以及紫花苜蓿的分布密度(478个/Mb).Varshney等[20 ] 研究认为,SSR分布密度之所以出现差异,除了物种间差异因素外,还与测序数据深度、序列拼接数据质量及SSR位点查找软件以及SSR搜索标准不同有关.研究选用主流的微卫星筛选软件MISA(MIcroSAtellite identification tool),在相同设置条件下分析了这3种苜蓿间差异.金花菜、蒺藜苜蓿和紫花苜蓿的测序深度分别为117X、109X和153X,均为二代+三代测序组装的高质量基因组,结果比较能真实发映出物种间的差异.金花菜基因组SSR的分布密度较低,可能与金花菜染色体数目少有关. ...
Conserved sequence preference in DNA binding among recombination proteins: An effect of ssDNA secondary structure
1
1999
... 单核苷酸、二核苷酸和三核苷酸重复单元是绝大多植物基因组SSR序列中优势重复单元[18 ,21 ] .研究发现,金花菜基因组SSR中,单、二和三核苷酸重复单元类型分别占基因组SSR数量的75.58%、15.31%和7.94%,其后依次是,四、五和六核苷酸重复单元,与蒺藜苜蓿观测到的结果相一致.与金花菜和蒺藜苜蓿相比,紫花苜蓿的六核苷酸重复单元数量多于五核苷酸重复单元,且单核苷酸重复单元类型的SSR数量相对较少,这可能与紫花苜蓿是同源四倍体,异花授粉导致遗传变异更为丰富有关. ...




{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}